Refinements modules
[emref] module
Energy minimization refinement with CNS.
The [emref] module refines the input structure or a complex by energy minimization using
the conjugate gradient method implemented in CNS.
Coordinates of the energy-minimized structures are saved, and each structure/complex is then evaluated using HADDOCK scoring function.
The default HADDOCK scoring function in the [emref] module is the following:
Notable parameters
The most important parameters for the [emref] module are:
ambig_fname: file containing the ambiguous interaction restraints (AIRs, optional)unambig_fname: file containing the unambiguous interaction restraints (optional)randremoval: whether or not to activate the random removal of restraints (default: True)nemsteps: number of energy minimization steps (default: 200)
More information about the [emref] parameters is available here or retrieved by running:
haddock3-cfg -m emref
[flexref] module
Flexible refinement with CNS.
The [flexref] module (previously known as it1 stage in HADDOCK2.X series),
is a semi-flexible simulated annealing (SA) protocol based on molecular
dynamics (MD) in torsion angle space.
This semi-flexible SA consists of four sequential stages:
- High-temperature rigid body MD
- Rigid body SA
- Semi-flexible SA with flexible side-chains at the interface
- Semi-flexible SA with fully flexible interface (both backbone and side-chains)
By default, only the interface regions are treated as semi-flexible. These regions are automatically defined based on intermolecular contacts. However, the user has the option to manually specify semi-flexible regions, and also define fully flexible regions that remain flexible throughout the entire protocol, starting from the high-temperature rigid-body MD stage.
See animation of the `[flexref]` protocol in action:
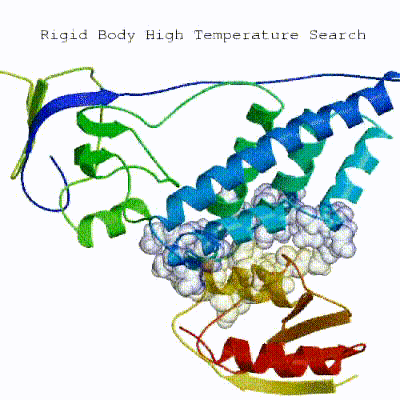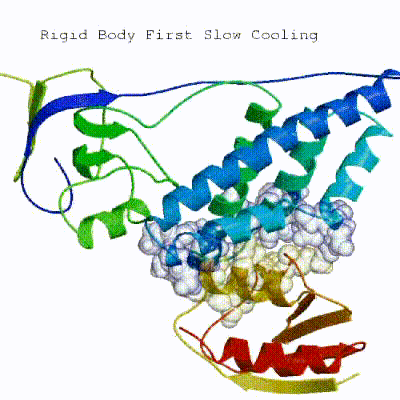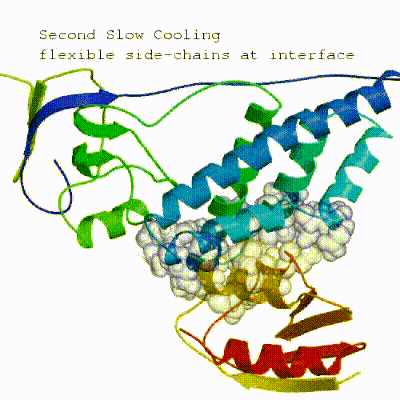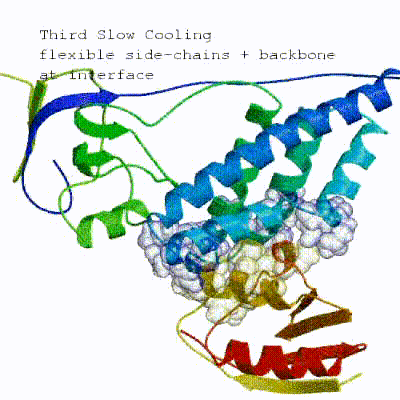
Here is a schematic visualization of the [flexref] stages with relevant parameters:
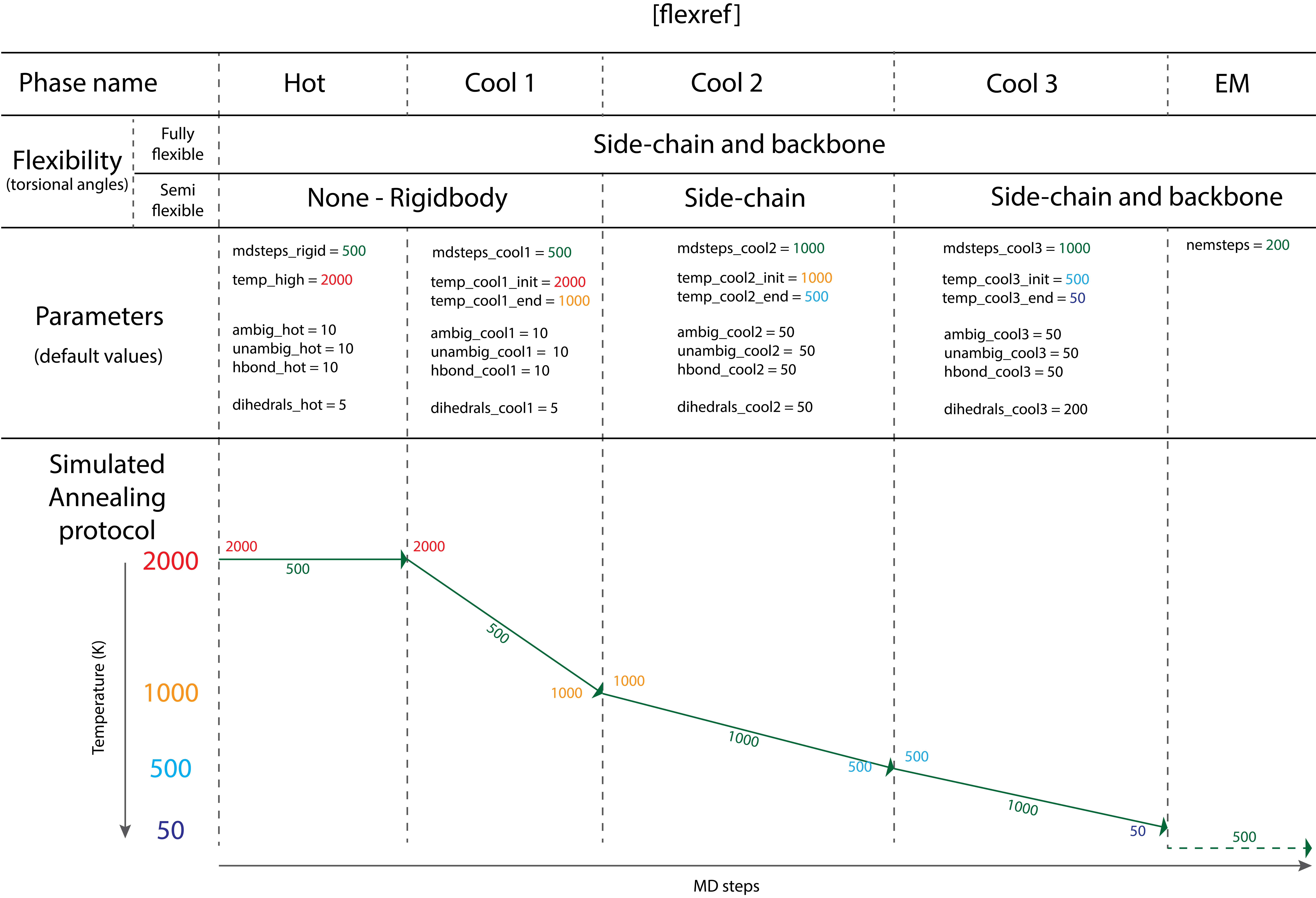
The temperature and number of steps for the various stages can be tuned.
The default HADDOCK scoring function in the [flexref] module is the following:
Notable parameters
The most important parameters for the [flexref] module are:
ambig_fname: file containing the ambiguous interaction restraints (AIRs, optional)unambig_fname: file containing the unambiguous interaction restraints (optional)seg_*_X_Y: for the definition of semi-flexible segments (see flexibility section for more information)fle_*_Y: for the definition of fully flexible segments (see flexibility section for more information)
More information about the [flexref] parameters is available here or retrieved by running:
haddock3-cfg -m flexref
[mdref] module
Explicit solvent MD refinement with CNS.
The [mdref] module (previously known as itw in HADDOCK2.X series), is a small MD simulation in cartesian space using explicit solvent.
A layer of solvent (8Å for water, 12.5Å for DMSO) is generated around surface residues.
The [mdref] protocol consists of four sequential steps:
- Short energy minimization
- Heating: 3 stages of short MD to reach the temperature of 300K (gradually increases the temperature, performing MD at 100K, 200K, and finally 300K)
- MD at 300K
- Cooling: 3 stages of short MD to reach the temperature of 100K (gradually decreases the temperature, performing MD at 300K, 200K, and finally 100K)
See animation of the `[mdref]` protocol in action:
Here is a schematic visualization of the [mdref] stages with relevant parameters:
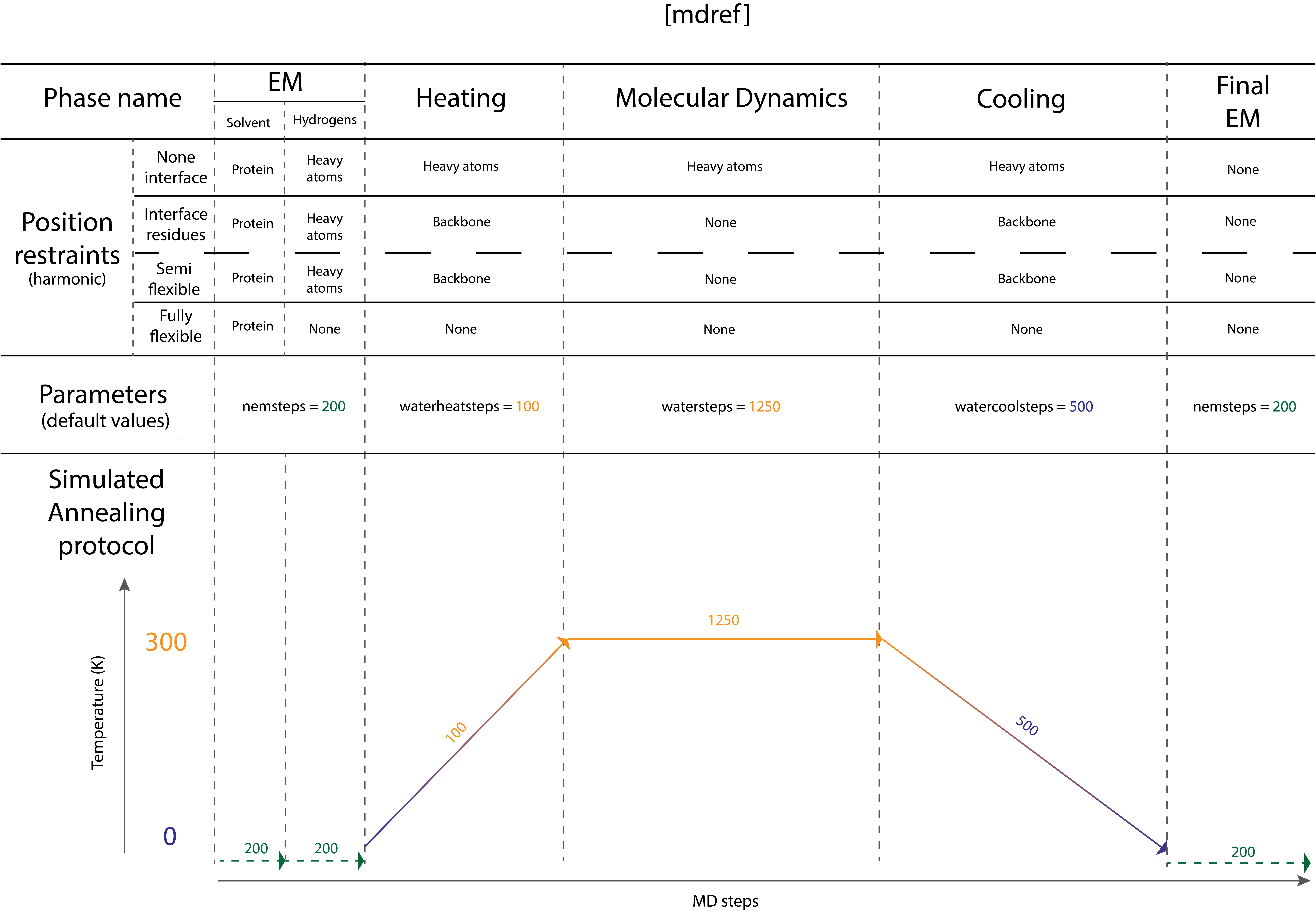
Using this protocol with default parameters, no spectacular changes are expected;
however, the scoring of the various structures may be improved.
The default HADDOCK scoring function in the [mdref] module is the following:
Notable parameters
The most important parameters for the [mdref] module are:
-
ambig_fname: file containing the ambiguous interaction restraints (AIRs, optional) -
unambig_fname: file containing the unambiguous interaction restraints (optional) -
waterheatsteps: number of MD steps for heating up the system (default: 100) -
watersteps: number of MD steps at 300K (default: 1250) -
watercoolsteps: number of MD steps for cooling down the system (default: 500)
More information about [mdref] parameters is available here or retrieved by running:
haddock3-cfg -m mdref
[cgtoaa] module
Backmapping of Coarse-Grained (CG) models into their All-Atom (AA) counterpart.
The [cgtoaa] module performs the backmapping of the CG representation into AA.
The back-mapping procedure uses the distance restraints (*_cg_to_aa.tbl) that were automatically generated by [topocg] module and propagated all along the workflow.
During the initial CG mapping stage in [topocg], atoms-to-bead restraints are defined, consisting of one distance restraint per bead (upper limit of 0Å) between the geometric center of the atoms and the bead to which they belong.
These restraints are used to morph the starting respective AA single structures onto the CG modelled complex.
This back-mapping protocol consists of three main steps:
- An initial fitting of the single AA structures onto their CG counterparts by restrained energy minimisation using the atoms-to-bead distance restraints, downscaling the intermolecular interactions to avoid high energies due to clashes between molecules;
- short cycles of restrained minimization and molecular dynamics introducing conformational changes into the AA structures to match the conformation of the CG model, gradually increasing the interaction strength between molecules and;
- final optimisation and clash removal through additional rounds of minimization and brief MD.
During all those steps, the CG model is kept rigid and fixed in space. Both AA and CG models co-exist, but don’t see each other except through the effect of the CG to AA distance restraints. At the end of the protocol, the CG model is deleted.
More information about the [cgtoaa] parameters is available here or retrieved by running:
haddock3-cfg -m cgtoaa
[openmm] module
The [openmm] modules makes use of the OpenMM molecular dynamics engine to perform the refinement of input structures (complexes or not).
The potential of OpenMM can be exploited to perform different tasks, such as:
-
Run MD simulation for each model from previous step;
-
Refine the models in the middle of a docking run. For example, it can be used to refine the models coming from a
[rigidbody]module before[flexref]is executed, or to replace the[mdref]step. -
Generate conformers prior to their use in a thorough docking run.
Here is a short description of the module's workflow:
-
Generate openmm topology and fix atoms
-
Build solvation box (water + ions) arround input structure
-
Equilibration solvation box restraining the protein heavy atoms
-
Run MD simulation: increase temperature, run MD, reduce temperature.
-
Either generate an ensemble of multiple frames or return the last frame.
Note that this module:
-
cannot make use of ambiguous restraints.
-
will refine all models coming from the previous workflow step and send them to the next step in the workflow. If you want to use other modules such as
[flexref]or[emref]after the[openmm]module, you need to recreate the topologies by simply adding a[topoaa]step in the workflow.
We provide some examples on our GitHub repository: haddock3 examples/thirdparty/openmm.
Notable parameters
The most important parameters for the [openmm] module are:
forcefield: Select the force-field in which to perform the simultation.simulation_timesteps: Number of MD simulation timesteps to perform.save_intermediate: Number of intermediate configurations to save during the simulation. The code divides the length of the simulation (simulation_timesteps) by this parameter to get how often frames should be saved.generate_ensemble: If 'true', generates 1 single ensemble file containing various frames; composed of the equilibrated one, all intermediates and the final one. If 'false', only return the last configuration obtained after 'simulation_timesteps' steps.sampling_factor: Number of simulation replicas for each input structure.
More information about [openmm] parameters is available here or retrieved by running:
haddock3-cfg -m openmm