Prepare Electron Microscopy Data¶
The powerfit program requires a EM density of a unknown structure where it can fit structures into.
Most of the the entries on EMDB contains multiple structures, so we want to pretent that we do not know one of structures.
For this example we will use EMD-33292, a sodium channel, with fitted model 7xm9.
The fitted model consist of following chains:
- A: Isoform 3 of Sodium channel protein type 9 subunit alpha
- B: Sodium channel subunit beta-1
- C: Sodium channel subunit beta-2
We will use the B chain, the sodium channel subunit beta-1, as the unknown structure.
Lets start by downloading the density map and the fitted model.
!wget -nc https://ftp.ebi.ac.uk/pub/databases/emdb/structures/EMD-33292/map/emd_33292.map.gz
!gunzip -kf emd_33292.map.gz
!wget -nc https://www.ebi.ac.uk/pdbe/entry-files/download/7xm9.cif
--2025-07-10 15:24:27-- https://ftp.ebi.ac.uk/pub/databases/emdb/structures/EMD-33292/map/emd_33292.map.gz Resolving ftp.ebi.ac.uk (ftp.ebi.ac.uk)... 193.62.193.165 Connecting to ftp.ebi.ac.uk (ftp.ebi.ac.uk)|193.62.193.165|:443... connected. HTTP request sent, awaiting response... 200 OK Length: 62515467 (60M) [application/x-gzip] Saving to: ‘emd_33292.map.gz’ emd_33292.map.gz 100%[===================>] 59.62M 75.4MB/s in 0.8s 2025-07-10 15:24:28 (75.4 MB/s) - ‘emd_33292.map.gz’ saved [62515467/62515467] File ‘7xm9.cif’ already there; not retrieving.
from pathlib import Path
in_density = Path("emd_33292.map")
pdb = Path("7xm9.cif")
unknown_chain = "B"
resolution = 3.48
masked_density = Path(f"{in_density.stem}-{pdb.stem}-{unknown_chain}-{resolution}.mrc")
script = masked_density.with_suffix(".cxc")
script.write_text(f"""\
open {in_density};
open {pdb};
delete #2/{unknown_chain};
molmap #2 {resolution};
volume mask #1 surfaces #3 invertMask true;
save {masked_density} #4;
exit
""")
148
# Because sodium channel is in the membrane the EM density has a donut of small density around the protein.
# This make fitting slow, because powerfit has to try to fit the template in the membrane density as well.
# We can cheat by taking just density of chain B with a bit of padding around it.
# This works when you want to try powerfit on a fitted model, but if you do not know the structure you want to fit, you can not do this.
from pathlib import Path
in_density = Path("emd_33292.map")
pdb = Path("7xm9.cif")
unknown_chain = "B"
resolution = 3.48
masked_density = Path(f"{in_density.stem}-{pdb.stem}-{unknown_chain}-{resolution}.cheated.mrc")
script = masked_density.with_suffix(".cxc")
script.write_text(f"""\
open {in_density};
open {pdb};
molmap #2/{unknown_chain} {resolution} balls true;
volume mask #1 surfaces #3 pad 4;
save {masked_density} #4;
exit
""")
146
!chimerax --nogui --script $script
Executing: runscript emd_33292-7xm9-B-3.48.cheated.cxc Executing: open emd_33292.map Computing emd_33292.map surface, level 0.293 Computing emd_33292.map surface, level 0.293 Calculated emd_33292.map surface, level 0.293, with 805008 triangles Opened emd_33292.map as #1, grid size 256,256,256, pixel 1.04, shown at level 0.293, step 1, values float32 Calculated emd_33292.map surface, level 0.293, with 805008 triangles Opened emd_33292.map as #1, grid size 256,256,256, pixel 1.04, shown at level 0.293, step 1, values float32 Executing: open 7xm9.cif _7xm9.cif_ title: **Cryo-EM structure of human NaV1.7/beta1/beta2-XEN907** [[more info...]](cxcmd:log metadata #2) Executing: open 7xm9.cif _7xm9.cif_ title: **Cryo-EM structure of human NaV1.7/beta1/beta2-XEN907** [[more info...]](cxcmd:log metadata #2) Chain information for 7xm9.cif #2 --- Chain | Description | UniProt [A](cxcmd:select /A:7-1769 "Select chain") | [Isoform 3 of Sodium channel protein type 9 subunit alpha,Green fluorescent protein](cxcmd:sequence chain #2/A "Show sequence") | [SCN9A_HUMAN](cxcmd:open Q15858 from uniprot associate #2/A "Show annotations") [1-1988](cxcmd:select #2/A:1-1988 "Select sequence") [B](cxcmd:select /B:20-192 "Select chain") | [Sodium channel subunit beta-1,Green fluorescent protein](cxcmd:sequence chain #2/B "Show sequence") | [SCN1B_HUMAN](cxcmd:open Q07699 from uniprot associate #2/B "Show annotations") [1-218](cxcmd:select #2/B:1-218 "Select sequence") [C](cxcmd:select /C:29-148 "Select chain") | [Sodium channel subunit beta-2](cxcmd:sequence chain #2/C "Show sequence") | [SCN2B_HUMAN](cxcmd:open O60939 from uniprot associate #2/C "Show annotations") [1-215](cxcmd:select #2/C:1-215 "Select sequence") Non-standard residues in 7xm9.cif #2 --- [6OU](cxcmd:sel :6OU "select residue") — [[(2~{R})-1-[2-azanylethoxy(oxidanyl)phosphoryl]oxy-3-hexadecanoyloxy- propan-2-yl] (~{Z})-octadec-9-enoate](http://www.rcsb.org/ligand/6OU "show residue info") [G2E](cxcmd:sel :G2E "select residue") — [(7~{R})-1'-pentylspiro[6~{H}-furo[3,2-f][1,3]benzodioxole-7,3'-indole]-2'-one](http://www.rcsb.org/ligand/G2E "show residue info") [NAG](cxcmd:sel :NAG "select residue") — [2-acetamido-2-deoxy-beta-D- glucopyranose](http://www.rcsb.org/ligand/NAG "show residue info") (N-acetyl- beta-D-glucosamine; 2-acetamido-2-deoxy-beta-D-glucose; 2-acetamido-2-deoxy-D- glucose; 2-acetamido-2-deoxy-glucose; N-ACETYL-D-GLUCOSAMINE) Executing: molmap #2/B 3.48 balls true Opened 7xm9.cif map 3.48 as #3, grid size 61,70,94, pixel 1.16, shown at level 0.636, step 1, values float32 Executing: volume mask #1 surfaces #3 pad 4 Chain information for 7xm9.cif #2 --- Chain | Description | UniProt [A](cxcmd:select /A:7-1769 "Select chain") | [Isoform 3 of Sodium channel protein type 9 subunit alpha,Green fluorescent protein](cxcmd:sequence chain #2/A "Show sequence") | [SCN9A_HUMAN](cxcmd:open Q15858 from uniprot associate #2/A "Show annotations") [1-1988](cxcmd:select #2/A:1-1988 "Select sequence") [B](cxcmd:select /B:20-192 "Select chain") | [Sodium channel subunit beta-1,Green fluorescent protein](cxcmd:sequence chain #2/B "Show sequence") | [SCN1B_HUMAN](cxcmd:open Q07699 from uniprot associate #2/B "Show annotations") [1-218](cxcmd:select #2/B:1-218 "Select sequence") [C](cxcmd:select /C:29-148 "Select chain") | [Sodium channel subunit beta-2](cxcmd:sequence chain #2/C "Show sequence") | [SCN2B_HUMAN](cxcmd:open O60939 from uniprot associate #2/C "Show annotations") [1-215](cxcmd:select #2/C:1-215 "Select sequence") Non-standard residues in 7xm9.cif #2 --- [6OU](cxcmd:sel :6OU "select residue") — [[(2~{R})-1-[2-azanylethoxy(oxidanyl)phosphoryl]oxy-3-hexadecanoyloxy- propan-2-yl] (~{Z})-octadec-9-enoate](http://www.rcsb.org/ligand/6OU "show residue info") [G2E](cxcmd:sel :G2E "select residue") — [(7~{R})-1'-pentylspiro[6~{H}-furo[3,2-f][1,3]benzodioxole-7,3'-indole]-2'-one](http://www.rcsb.org/ligand/G2E "show residue info") [NAG](cxcmd:sel :NAG "select residue") — [2-acetamido-2-deoxy-beta-D- glucopyranose](http://www.rcsb.org/ligand/NAG "show residue info") (N-acetyl- beta-D-glucosamine; 2-acetamido-2-deoxy-beta-D-glucose; 2-acetamido-2-deoxy-D- glucose; 2-acetamido-2-deoxy-glucose; N-ACETYL-D-GLUCOSAMINE) Executing: molmap #2/B 3.48 balls true Opened 7xm9.cif map 3.48 as #3, grid size 61,70,94, pixel 1.16, shown at level 0.636, step 1, values float32 Executing: volume mask #1 surfaces #3 pad 4 Opened emd_33292.map masked as #4, grid size 61,71,99, pixel 1.04, shown at step 1, values float32 Executing: save emd_33292-7xm9-B-3.48.cheated.mrc #4 Wrote file emd_33292-7xm9-B-3.48.cheated.mrc Executing: exit Exiting ... Opened emd_33292.map masked as #4, grid size 61,71,99, pixel 1.04, shown at step 1, values float32 Executing: save emd_33292-7xm9-B-3.48.cheated.mrc #4 Wrote file emd_33292-7xm9-B-3.48.cheated.mrc Executing: exit Exiting ...
# Clean after ourselves
script.unlink()
masked_density, pdb
(PosixPath('emd_33292-7xm9-B-3.48.cheated.mrc'), PosixPath('7xm9.cif'))
Open the masked_density file in a viewer to verify that densities of chain A and C from pdb file have been removed.
To check that the created density can be used by powerfit we can run powerfit with it.
Create a re-oriented template structure¶
We could just fit chain B from 7xm9.cif, but as it is already in the correct position and orientation this feels a bit like cheating. we can make a new template structure that has incorrect position and orientation.
Let us use gemmi to apply a rotation and translation to the B chain of 7xm9.cif.
import math
import gemmi
def create_rotation_matrix(rotations):
result = gemmi.Mat33()
for axis in ["z", "y", "x"]:
if axis in rotations:
angle = rotations[axis]
c, s = math.cos(angle), math.sin(angle)
if axis == "x":
matrix = gemmi.Mat33([[1, 0, 0], [0, c, -s], [0, s, c]])
elif axis == "y":
matrix = gemmi.Mat33([[c, 0, s], [0, 1, 0], [-s, 0, c]])
else: # axis == "z"
matrix = gemmi.Mat33([[c, -s, 0], [s, c, 0], [0, 0, 1]])
result = matrix.multiply(result)
return result
transform = gemmi.Transform(mat33=create_rotation_matrix({"x": 0.5, "y": 0.1, "z": 0.2}), vec3=gemmi.Vec3(40, 50, 60))
structure = gemmi.read_structure(str(pdb))
gemmi.Selection(unknown_chain).remove_not_selected(structure)
model = structure[0]
model.transform_pos_and_adp(transform)
# Save the modified structure
template = pdb.with_suffix(".B-reoriented.cif")
doc = structure.make_mmcif_document(gemmi.MmcifOutputGroups(True, chem_comp=False))
doc.write_file(str(template))
print(f"Saved reoriented chain {unknown_chain} to {template}")
Saved reoriented chain B to 7xm9.B-reoriented.cif
Fit with powerfit¶
powerfit_result_dir = Path(f"powerfit-{masked_density.stem}")
!powerfit $masked_density $resolution $template -d $powerfit_result_dir --delimiter , -p 6
Target file read from: /home/verhoes/git/protein-detective/protein-detective/docs/emd_33292-7xm9-B-3.48 .cheated.mrc Target resolution: 3.48 Initial shape of density: 99 71 61 Shape after trimming: 94 71 59 Shape after extending: 96 72 60 Template file read from: /home/verhoes/git/protein-detective/protein-detective/docs/7xm9.B-reoriented.cif Calculating core-weighted mask. Reading in rotations. Requested rotational sampling density: 10.00 Real rotational sampling density: 9.72 Requested number of processors: 6 Starting search Processing rotations 100% ━━━━━━━━━╸ 7,415/7,… [ 0:00:32 < 0:00:01 , 232 rot/s ]232 rot/s ] Processing rotations 100% ━━━━━━━━━╸ 7,415/7,… [ 0:00:32 < 0:00:01 , 232 rot/s ] Time for search: 0m 32s Analyzing results Time for search: 0m 32s Analyzing results Writing solutions to file. Writing PDBs to file. Writing solutions to file. Writing PDBs to file. Total time: 0m 32s Total time: 0m 32s
powerfit_results = powerfit_result_dir / "solutions.out"
!head $powerfit_results
rank,cc,Fish-z,rel-z,x,y,z,a11,a12,a13,a21,a22,a23,a31,a32,a33 1,0.161,0.162,10.737,122.720,101.920,170.560,0.978,0.109,0.176,-0.176,0.886,0.429,-0.109,-0.450,0.886 2,0.093,0.093,6.175,122.720,104.000,167.440,0.891,0.452,-0.038,-0.388,0.803,0.452,0.235,-0.388,0.891 3,0.089,0.090,5.942,126.880,98.800,172.640,1.000,-0.000,0.000,0.000,0.815,0.580,-0.000,-0.580,0.815 4,0.089,0.089,5.911,112.320,81.120,171.600,0.000,0.580,-0.815,1.000,0.000,0.000,0.000,-0.815,-0.580 5,0.088,0.089,5.866,119.600,104.000,174.720,0.918,0.182,0.353,-0.353,0.781,0.515,-0.182,-0.597,0.781 6,0.088,0.088,5.813,121.680,105.040,166.400,0.891,0.452,-0.038,-0.388,0.803,0.452,0.235,-0.388,0.891 7,0.086,0.086,5.701,137.280,92.560,168.480,0.294,0.294,-0.910,-0.045,0.955,0.294,0.955,-0.045,0.294 8,0.085,0.085,5.622,113.360,104.000,182.000,-0.216,0.517,0.828,0.729,0.649,-0.216,-0.649,0.557,-0.517 9,0.083,0.084,5.541,128.960,92.560,168.480,0.452,0.388,-0.803,0.038,0.891,0.452,0.891,-0.235,0.388
Visualize fit¶
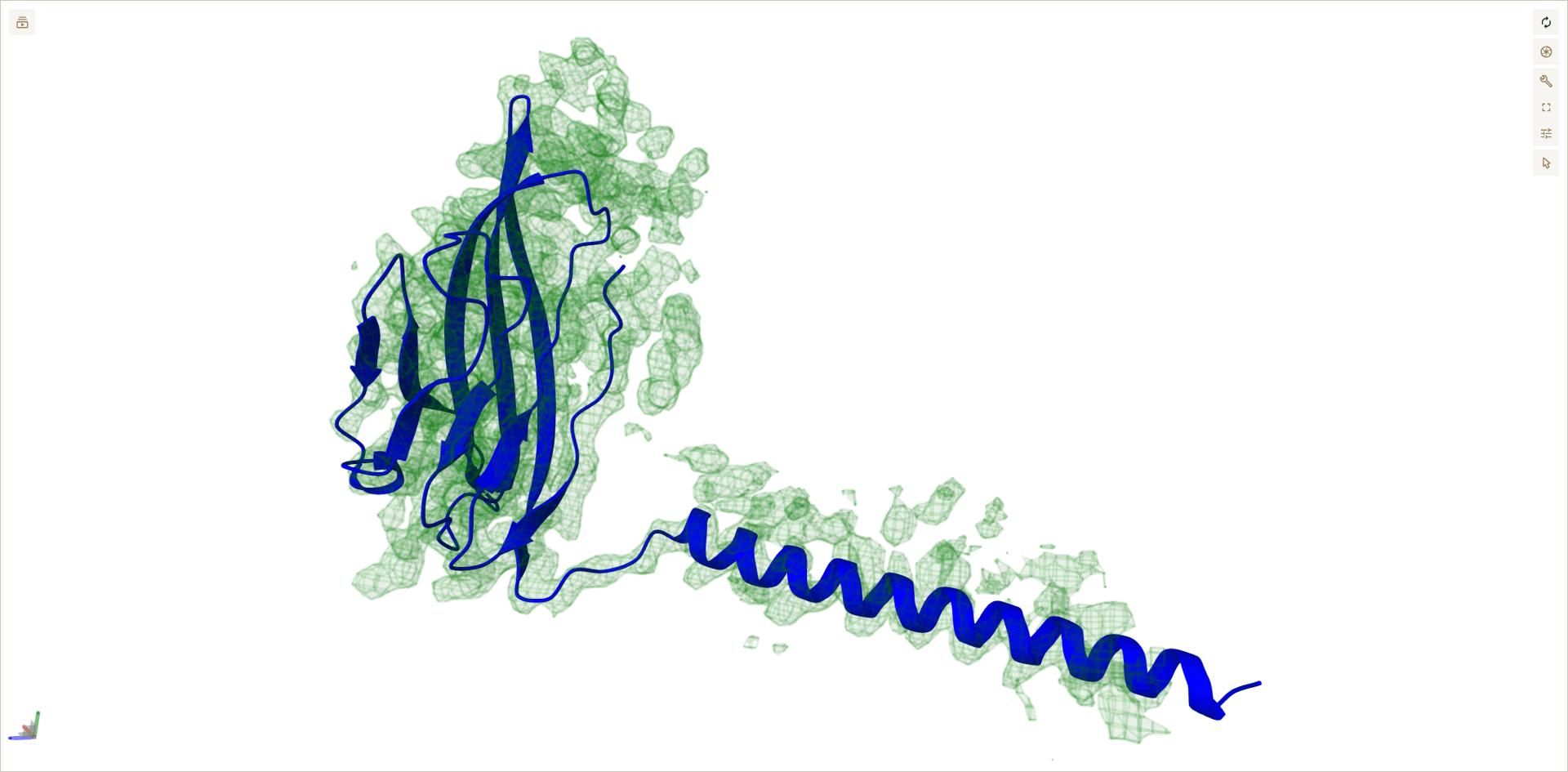
# To visualize interactively run this cell
from protein_detective.visualization import show_structure_and_density
best_fit = powerfit_result_dir / "fit_1.pdb"
show_structure_and_density(best_fit, masked_density)