

HADDOCK small molecule binding site screening
Introduction
In this tutorial we will use HADDOCK in its ab-initio mode to try to identify putative binding sites for small ligands on a protein receptor, using as example the multidrug exporter AcrB, described in the following publication:
Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors.
Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG
PLoS Biol. 5 e7 (2007)
ABSTRACT: “The multidrug exporter AcrB is the inner membrane component of the AcrAB-TolC drug efflux system in Escherichia coli and is responsible for the resistance of this organism to a wide range of drugs. … The three subunits of AcrB are locked in different conformations revealing distinct channels in each subunit. There seems to be remote conformational coupling between the channel access, exit, and the putative proton-translocation site, explaining how the proton motive force is used for drug export. Thus our structure suggests a transport pathway not through the central pore but through the identified channels in the individual subunits, which greatly advances our understanding of the multidrug export mechanism.”
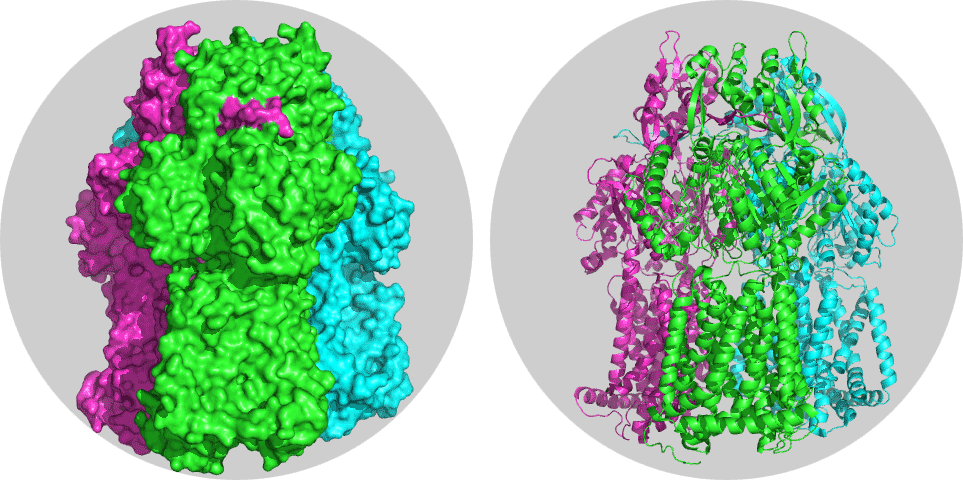
This tutorial consists of the following sections:
- Introduction
- Setup
- Inspecting the content of the tutorial
- Preparing PDB files of the receptor for docking
- Preparing PDB files of the ligands for docking
- Ab-initio surface-based docking with HADDOCK
- First analysis of the results
- Statistical contact analysis
- Identifying a binding pocket from the contact statistics
- Setting up a new docking run targeting the identified binding pocket
- Analysis of the targeted docking results
In the first part of this tutorial you will learn to clean and manipulate PDB files in preparation for docking. Then we will setup an ab-initio docking run in HADDOCK using surface restraints randomly selected from all accessible residues in order to sample the entire surface of the receptor (the so-called surface contact restraints in HADDOCK). A statistical analysis of the docking models in terms of most contacted residues will then be performed to identify and visualize putative binding sites. Finally, the results from this statistical analysis will be used to setup a protein-ligand docking run targeting the predicted binding sites.
For this tutorial we will make use of the HADDOCK2.2 webserver. A description of our web server can be found in the following publications:
-
G.C.P van Zundert, J.P.G.L.M. Rodrigues, M. Trellet, C. Schmitz, P.L. Kastritis, E. Karaca, A.S.J. Melquiond, M. van Dijk, S.J. de Vries and A.M.J.J. Bonvin. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol., 428, 720-725 (2015).
-
S.J. de Vries, M. van Dijk and A.M.J.J. Bonvin. The HADDOCK web server for data-driven biomolecular docking. Nature Protocols, 5, 883-897 (2010). Download an open version here.
Throughout the tutorial, coloured text will be used to refer to questions or instructions, Linux and/or Pymol commands.
This is a question prompt: try answering it! This an instruction prompt: follow it! This is a Pymol prompt: write this in the Pymol command line prompt! This is a Linux prompt: insert the commands in the terminal!
Setup
In order to run this tutorial you will need to have Pymol installed. You can of course use instead your favorite structure viewer, but the visualization commands described here are for Pymol.
Further you should install our PDB-tools, or clone it from the command line:
git clone https://github.com/haddocking/pdb-tools
Make sure that the pdb-tools directory is in your search path. For this go into the pdb-tools directory and then if working under tsch type:
And for bash:
Download then the data to run this tutorial from our GitHub data repository here or clone it from the command line:
git clone https://github.com/haddocking/HADDOCK-binding-sites-tutorial
Alternatively, if you do not have git installed, simply go the above web address and download the zip archive.
You will also need to compile a few provided programs for post-analysis.
For this go into the ana_scripts directory of the cloned directory and type make
(we are here assuming a tcsh or csh shell):
cd HADDOCK-binding-sites-tutorial/ana_scripts
make
source setup.csh
cd ..
Note: This is defining some environment variable which we will use in the following. Repeat this step and the above step about defining the path everytime you open a new terminal window.
If you don’t want to wait with the docking runs to complete in order to proceed with the analysis (see section about Preparing PDB files for docking section below), you can already download pre-calculated runs using the script provided into the runs directory:
cd runs
./download-run-data.csh
cd ..
This will download two reduced docking runs, one for the random sampling of the surface and one for the targeted protein-ligand docking (about 450MB of compressed data).
Or to download two full docking runs, one for the random sampling of the surface and one for the targeted protein-ligand docking (about 6GB of compressed data).:
cd runs
./download-run-data-full.csh
cd ..
Inspecting the content of the tutorial
Let us first inspect the various files provided with this tutorial. You will see three directories and one file:
-
HADDOCK-runfiles: this directory contains the reference HADDOCK parameter files for various docking runs described in this tutorial. These can be used to reproduce the docking using the file upload interface of the HADDOCK server.
-
ana_scripts: this directory contains various analysis scripts to analyse the results of the docking, including the statistical contact analysis.
-
pdbs: this directory contains various PDB files which will be used during this tutorial
-
pdbs-processed: this directory contains processed, cleaned PDB files (See the Preparing PDB files for docking section below), ready for docking
-
runs: this directory contains scripts that allows you to download pre-calculated docking runs.
Preparing PDB files of the receptor for docking
One requirement of HADDOCK is that there should not be any overlap in residue numbering. The structure of the apo form of our target receptor, the multidrug efflux pump AcrB from Escherichia coli, is available from the Protein Data Bank under PDB ID 2J8S. You can download it directly from the PDB using the pdb_fetch.py script from our pdb-tools utilities:
The file is also provided in the pdbs directory.
Let’s first inspect this structure using Pymol (or your favorite viewer):
And in pymol type at the prompt level:
Take some time to inspect the 3D structure. Each chain should have a different color.
How many chains can you identify?
If you look at the desciption of this structure on the PDB website, it states “DRUG EXPORT PATHWAY OF MULTIDRUG EXPORTER ACRB REVEALED BY DARPIN INHIBITORS”. You should be able to identify the two darpins (they have chainIDs D and E in the structure). Let’s remove them in pymol:
Now you only see the mutlidrug exporter. It consists of chain A,B and C and is the system which we will further use for docking, but we have to make sure first that there is no overlap in numbering.
For this we will work at the terminal level and use our pbd-tools utilities. Quit first pymol.
Let’s first find out what are the first and last residue numbers of the various chains, to check if there is any overlap in numbering:
pdb_selchain.py -A 2J8S.pdb | grep ' CA ' | grep ATOM | head -5
pdb_selchain.py -A 2J8S.pdb | grep ' CA ' | grep ATOM | tail -5
pdb_selchain.py -B 2J8S.pdb | grep ' CA ' | grep ATOM | head -5
pdb_selchain.py -B 2J8S.pdb | grep ' CA ' | grep ATOM | tail -5
pdb_selchain.py -C 2J8S.pdb | grep ' CA ' | grep ATOM | head -5
pdb_selchain.py -C 2J8S.pdb | grep ' CA ' | grep ATOM | tail -5
Inspecting the results of those commands reveals that we are indeed dealing with overlapping numbering: all three chains start at residue 1.
For use in HADDOCK we have thus to renumber chain B and C. In order to easily match the residue numbers between chains it is advisable to shift the numbering by a round number, e.g. in this case since we have more than 1000 amino acids we can shift chain B and C by 2000 and 4000, respectively. We will use again our pdb-tools utilities to create a renumbered, clean PDB file (also removing all hetero atoms in the process by selection only ATOM recoords):
The PDB file of our receptor should now be ready for docking. You can also check the file format with:
pdb_validate.py 2J8S-renumbered.pdb
This will report formatting issues.
Preparing PDB files of the ligands for docking
Several small molecules are known to bind to this receptor, among which rifampicin and minocycline. A crystal structure of the complex with both ligands is also available from the PDB website (PBD entry 3AOD). Those ligands are binding to two different sites on the receptor.
For docking we need coordinates of those ligands in PDB format with line starting with HETATM. After downloading the corresponding PDB entry 3AOD extract the ligands from it with the following commands:
For rifampicin (called RFP in the PDB file):
grep RFP 3AOD.pdb |grep HETATM > rifampicin.pdb
echo END >> rifampicin.pdb
For minocycline (called MIY in the PDB file):
grep MIY 3AOD.pdb |grep HETATM > minocycline.pdb
echo END >> minocycline.pdb
Ab-initio surface-based docking with HADDOCK
We will launch here a docking run using the apo form of the receptor (the renumbered PDB we just prepared) and rifampicin as potential ligand. For this we will make use of the guru interface of the HADDOCK web server, which does require guru level access (provided with course credentials if given to you, otherwise register to the server and request this access level):
https://alcazar.science.uu.nl/services/HADDOCK2.2/haddockserver-guru.html
Note: The blue bars on the server can be folded/unfolded by clicking on the arrow on the right
-
Step1: Define a name for your docking run, e.g. AcrB-rifampicin-surface.
-
Step2: Input the protein PDB file. For this unfold the Molecule definition menu.
First molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (to select all three chains) PDB structure to submit -> Browse and select 2J8S-renumbered.pdb Segment ID to use during docking -> A The N-terminus of your protein is positively charged -> uncheck the box if needed The C-terminus of your protein is negatively charged -> uncheck the box if needed
(Our structure might not be the real fragment used for crystallisation - better to have uncharged termini)
- Step 3. Input the ligand PDB file. For this unfold the Molecule definition menu.
Second molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All PDB structure to submit -> Browse and select rifampicin.pdb Segment ID to use during docking -> B
- Step 4: Turn on random surface restraints. For this unfold the Distance restraints menu
Define randomly ambiguous interaction restraints from accessible residues -> Check the box
- Step 5: Since we are doing ab-initio docking we do need to increase the sampling. For this unfold the Sampling parameters menu:
Number of structures for rigid body docking -> 10000 Number of structures for semi-flexible refinement -> 400 Number of structures for the explicit solvent refinement -> 400
Note: If you use course credentials, these numbers will be reduced to 500/50/50 to save computing time and get back results faster. You can also manually decrease those numbers and download instead a full pre-calculated run for analysis (see setup above).
- Step 6: Change the clustering settings since we are dealing with a small molecule. For this unfold the Clustering parameter menu:
Clustering method (RMSD or Fraction of Common Contacts (FCC)) -> RMSD RMSD Cutoff for clustering (Recommended: 7.5A for RMSD, 0.75 for FCC) -> 2.0
- Step 7: Apply some ligand-specific scoring setting. For this unfold the Scoring parameter menu:
Our recommended HADDOCK score settings for small ligands docking are the following:
HADDOCKscore-it0 = 1.0 Evdw + 1.0 Eelec + 1.0 Edesol + 0.01 Eair - 0.01 BSA
HADDOCKscore-it1 = 1.0 Evdw + 1.0 Eelec + 1.0 Edesol + 0.1 Eair - 0.01 BSA
HADDOCKscore-water = 1.0 Evdw + 0.1 Eelec + 1.0 Edesol + 0.1 Eair
This differs from the defaults setting (defined for protein-protein complexes). We recommend to change two weights for protein-ligand docking:
- Step 8: Apply some ligand-specific protocol setting. For this unfold the Advanced sampling parameter menu:
initial temperature for second TAD cooling step with flexible side-chain at the inferface -> 500
initial temperature for third TAD cooling step with fully flexible interface -> 300
number of MD steps for rigid body high temperature TAD -> 0
number of MD steps during first rigid body cooling stage -> 0
- Step 8: You are ready to submit! Enter your username and password (or the course credentials provided to you). Remember that for this interface you do need guru access.
First analysis of the results
Once your run has completed (this can take quite some time considering the size of the receptor) you will be presented with a result page showing the cluster statistics and some graphical representation of the data. Such an example output page can be found here.
Instead, you can also use the precalculated run. Simply unpack the archive (see the Setup section for downloading the archives), go into the directory and open with your favorite web browser the index.html file to view the results page.
Considering the size of the receptor we are targeting, at this stage it is rather unlikey that any sensible results will be obtained. If you performed the docking with course credentials, most likely the run will have completed but the minimum number of structures per cluster will have automatically reduced to 2 or even 1 in order to produce a result page. If 1, then the clusters reported on the web page will correspond to the top10 ranked models.
You can download the full run as a gzipped tar archive and inspect the results. Copy for this the link provided in the result page and download the archive with:
curl -L -O <link> or wget <link>
Unpack the gzip file with:
Note: You can also view a result page from a downloaded pre-calculated docking run. For this go into the runs directory and then download the runs using:
This will download two docking runs performed under course settings (i.e. reduced number of models), for a total of about 450MB of compressed data. Unpack the runs (using tar xfz <archive>.tgz).
Those contain the same html result page that the server would be returning. To view those, open in your favourite browser the index.html file provided in the run directory.
If you want to inspect some of the docking models, change directly to runname/structures/it1/water/analysis. In that directory you will find the models numbered according to their HADDOCK ranking, e.g. complexfit_1.pdb, complexfit_2.pdb, ...
You can for example inspect the first 10 with pymol, comparing them to the reference complex 3AOD:
pymol complexfit_[1-9].pdb complexfit_10.pdb $WDIR/3AOD-renumbered.pdb
This will load the top 10 models and a renumbered reference crystal structure into pymol (make sure to have run the Setup described above). In case WDIR is not defined or you are not running under a linux-like environment you can find the reference structure into the ana_scripts directory.
Note: The chain nomenclature between the 3AOD and the 2J8S structures differ. Chain C,A,B of 3AOD actually correspond to chain A,B,C of 2J8S. The renumbered 3AOD-renumbered.pdb file has been renumbered in such a way that the chains now match.
In pymol you can type the following to superimpose the models and change the style:
Can you find any ligand (in orange) close to the position of any of the two ligands in the reference crystal structure (in red)? Also consider that the receptor consists of three identical chains, but in slighly difference conformations. There might thus be symmetry-related binding site.
Statistical contact analysis
We will now perform a statistical analysis of all residues making contacts with the ligand at the rigid body docking stage. For this, go first into the runname/structures/it0 directory.
We first calculate all intermolecular contacts within 5Å for all models with the following command:
$WDIR/contacts-analysis.csh `cat file.nam`
This creates a contacts directory containing the list of intermolecular atomic contacts for each model.
Then we simply count the number of times a residue is contacted:
$WDIR/contacts-statistics.csh `cat file.nam`
Note: If you want to only analyze for example the top 1000 ranked models use instead:
$WDIR/contacts-statistics.csh `head -1000 file.nam`
The script generates for each chain a sorted list of residue with their number of contacts (on a residue basis). E.g. for chain A:
6 700 A 6 693 A 6 692 A 6 4429 A 6 2226 A 5 965 A 5 811 A 5 708 A 5 532 A ...
We can encode the contacts statistics in the B-factor field of a PDB file to allow for visualisation. For this we should use a PDB file with chainIDs.
Go into our example run directory, i.e. the run we downloaded from the HADDOCK server called AcrB-rifampicin-surface.
First let’s put back the chainID information in one of the starting model taken from the begin directory and set all B-factors to 1:
pdb_segxchain.py begin/protein1.pdb | pdb_b.py -1 > AcrB_contacts.pdb
And then we will use this PDB file, together with the contacts statistics file just created in structures/it0 to encode the contacts into the b-factor column of the PDB file with the following command:
$WDIR/encode-contacts.csh structures/it0/Acontacts.lis AcrB_contacts.pdb
The result is a new PDB file called AcrB_contacts.pdb which can now be visualized in Pymol (we also load here the reference structure).
If you have performed the analysis on a full run, use the AcrB_contacts.pdb file you just created. Otherwise, in order to get more significant results,
use instead the model provided in the AcrB-rifampicin-surface-full directory in which you will find the pre-computed data from an analysis
of 10000 rigid body docking (it0 models). The corresponding full run archive can be downloaded using the download-run-data-full.csh script
(but beware it is a large amount of data >10GB when unpacked).
pymol AcrB_contacts.pdb $WDIR/3AOD-renumbered.pdb
And then in Pymol type:
The above commands will display the surface of the molecule colored according to the contact frequency (from red 100 to blue 1). Also visible are the two ligands in the reference structure of the complex.
Note: If you want to put more emphasis on the most contacted regions, change the minimum value in the above command to for example 50.
Inspect the surface: Are there any highly contacted regions close to the actual binding sites?
Remember here that the receptor consists of three identical chains. For the docking we renumbered the chain to avoid overlap and gave them a unique segid (A). You can distinguish the various chains by the corresponding resisude numbering:
- Chain A starts at residue number 1
- Chain B starts at residue number 2001
- Chain C starts at residue number 4001.
The original paper by Sennhauser et al indicates that the three chains are locked in different conformations. They report in particular that chain B has the largest channel opening in their structure:
“The three AcrB subunits are bound in three different conformations, revealing three distinct channels (Figure 3). The width of these channels is sufficient for the passage of typical AcrB substrates. In subunit A, a channel is observed, extending from the external depression through the large periplasmic domain reaching almost the central funnel at the top of the protein (Figure 4A). Here the side chains of residues Gln124, Gln125, and Tyr758 form a gate, closing the channel and therefore preventing direct access to the central funnel. … A similar channel, although a little wider, is present in subunit B (Figure 4B). In addition, the channel is open not only to the periplasm but also to the membrane bilayer at the periphery of the TM domain. In subunit C, the channel entrances are closed due to movements of PC2 and PN1 (Figure 4C).”
To which one of the three channels/subunit do we observe preferential contacts on docking models?. Is this consistent with the observations by Sennhauser et al?.
Note: You can repeat the same analysis for a full docking run if you are interested. For this download the full run archives using the download-run-data-full.csh script in the runs directory (about 6GB of compressed data!). But instead you also only look at the encoded contacts statistics in the PDB file provided in the runs directory: AcrB-rifampicin-surface-full-contacts.pdb. This files contains the results of the contacts analysis of 10000 rigid body docking models.
pymol AcrB-rifampicin-surface-full-contacts.pdb $WDIR/3AOD-renumbered.pdb
And then in Pymol type:
Identifying a binding pocket from the contact statistics
We will now make use of the contact statistics obtained previously to target a specific binding site in a new docking run. We should make use of statistics obtained from a full docking run. In the previous section we have identified two preferred binding pockets in what should be chain A or chain B of the receptor. Chain B has a residue numbering which starts at 2001. In the renumbered 3AOD structure, rifampicin is found to bind to chain A.
We can extract the most contacted residue for chain B from the file containing the contacts statistics provided in the runs directory and called AcrB-rifampicin-surface-full-contacts.lis
So first change directory to the runs dir.
Let us first see how many residues are sampled in chain B and what are the highest and lowest number of contacts. The following command will give the total number of residues in chain A contacted by the ligand out of the 10000 models analysed:
awk '$2>2000 && $2<4000' AcrB-rifampicin-surface-full-contacts.lis |wc -l
Calculate what would correspond to the top 10% of the most contacted residues.
The most contacted residues are found at the head of this file:
awk '$2>2000 && $2<4000' AcrB-rifampicin-surface-full-contacts.lis |head -5
See solution:
198 2717 A 171 2566 A 166 2830 A 158 2715 A 154 2029 A
And the less often contacted residues at the bottom:
awk '$2>2000 && $2<4000' AcrB-rifampicin-surface-full-contacts.lis |tail -5
See solution:
1 2379 A 1 2351 A 1 2329 A 1 2200 A 1 2132 A
Let us now extract the list of the top 10% most contacted residues (change the value in the head statement if required) and store it to a new file:
We can now encode this information in a PDB file to visualize the defined binding site:
Load the resulting model in pymol and type:
zoom vis
hide lines
show cartoon
show mesh
spectrum b, blue_white_red, minimum=1, maximum=100
You should now be looking only at highly contacted regions of chain B. There is a clear binding pocket visible in the chainB corresponding to the entrance of the channel as described by Sennhauser et al (see Figure 3). This is the left view in the figure below. A second highly contacted region seems to be in the inside of the trimer (right picture below).
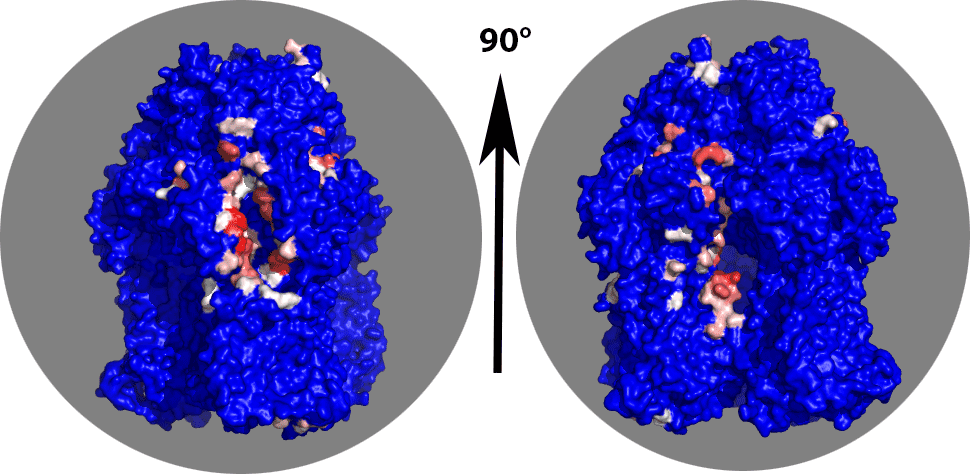
Try to figure out which residue numbers are lining the binding pocket shown on the left figure. From an analyis in Pymol it looks like these are mainly located between residue 2550 and 2725. We can extract their number from the top 10 contact statistics file with:
awk '$2>2550 && $2<2725' AcrB-rifampicin-surface-full-contacts-top10.lis | awk '{printf "%s, ", $2}'
See solution:
2717, 2566, 2715, 2722, 2676, 2562, 2677, 2678, 2645, 2561, 2580, 2690, 2694, 2662, 2579, 2674, 2675, 2689, 2718, 2664, 2560, 2667, 2564, 2693, 2716, 2700, 2666, 2563, 2565, 2554, 2577, 2601
Save this list since we will need it to setup the targeted docking run.
Setting up a new docking run targeting the identified binding pocket
We will now setup a second docking run targeting specifically the identified binding pocket on chain B. For our targeted ligand docking protocol, we will first create two sets of restraints which we will use at different stages of the docking:
-
For the rigid-body docking, we will first define the entire binding pocket on the receptor as active and the ligand as active too. This will ensure that the ligand is properly drawn inside the binding pocket.
-
For the subsequent flexible refinement stages, we define the binding pocket only as passive and the ligand as active. This ensures that the ligand can explore the binding pocket.
In order to create those two restraint files, use the Generate Restraints option from the haddock-restraints tool accessible at https://wenmr.science.uu.nl/haddock-restraints/:
Selection 1: Active residues (directly involved in the interaction) -> enter here the list of residues defining the binding site (see above) Selection 2: Active residues (directly involved in the interaction) -> enter here the residue number of our ligand rifampicin (2002) Click on submit and save the resulting page, naming it AcrB-rifampicin-act-act.tbl
Note: This works best with Firefox. Currently when using Chrome, saving as text writes the wrong info to file. In that case copy the content of the page and paste it in a text file.
Note: Avoid Safari for the time being - it is giving problems (we are working on it).
Now repeat the above steps, but this time entering the list of residues for the binding pocket into the passive residue list. Save the resulting restraint file as AcrB-rifampicin-pass-act.tbl
Note: Two pre-generated distance restraints files are available in the runs directory:
AcrB-rifampicin-act-act.tbl
AcrB-rifampicin-pass-act.tbl
The number of distance restraints defined in those file can be obtained by counting the number of times that an assign statement is found in the file, e.g.:
grep -i assign AcrB-rifampicin-act-act.tbl | wc -l
Compare the two generated files: what are the differences? How many restraints are defined in each?
Note: A description of the restraints format can be found in Box 4 of our Nature Protocol 2010 server paper:
- S.J. de Vries, M. van Dijk and A.M.J.J. Bonvin. The HADDOCK web server for data-driven biomolecular docking. Nature Protocols, 5, 883-897 (2010). Download an open version here.
We have now all the required information to setup our targeted docking run. We will again make use of the guru interface of the HADDOCK web server, which does require guru level access (provided with course credentials if given to you, otherwise register to the server and request this access level):
https://alcazar.science.uu.nl/services/HADDOCK2.2/haddockserver-guru.html
Note: The blue bars on the server can be folded/unfolded by clicking on the arrow on the right
-
Step1: Define a name for your docking run, e.g. AcrB-rifampicin-pocket.
-
Step2: Input the protein PDB file. For this unfold the Molecule definition menu.
First molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (to select all three chains) PDB structure to submit -> Browse and select 2J8S-renumbered.pdb Segment ID to use during docking -> A The N-terminus of your protein is positively charged -> uncheck the box if needed The C-terminus of your protein is negatively charged -> uncheck the box if needed
(Our structure might not be the real fragment used for crystallisation - better to have uncharged termini)
- Step 3. Input the ligand PDB file. For this unfold the Molecule definition menu.
Second molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All PDB structure to submit -> Browse and select rifampicin.pdb Segment ID to use during docking -> B
- Step 4: Input the restraint files for docking. For this unfold the Distance restraints menu
Instead of specifying active and passive residues, you can supply a HADDOCK restraints TBL file (ambiguous restraints) -> Upload here the AcrB-rifampicin-act-act.tbl You can supply a HADDOCK restraints TBL file with restraints that will always be enforced (unambiguous restraints) -> Upload here the AcrB-rifampicin-pass-act.tbl
- Step 5: Change the clustering settings since we are dealing with a small molecule. For this unfold the Clustering parameter menu:
Clustering method (RMSD or Fraction of Common Contacts (FCC)) -> RMSD RMSD Cutoff for clustering (Recommended: 7.5A for RMSD, 0.75 for FCC) -> 2.0
- Step 6: Define when to use each of the two restraint files we are uploading: For this unfold the Restraints energy constants menu”, and in that menu unfold the Energy constants for ambiguous restraints menu.
- Step 7: Apply some ligand-specific scoring setting. For this unfold the Scoring parameter menu:
Our recommended HADDOCK score settings for small ligands docking are the following:
HADDOCKscore-it0 = 1.0 Evdw + 1.0 Eelec + 1.0 Edesol + 0.01 Eair - 0.01 BSA
HADDOCKscore-it1 = 1.0 Evdw + 1.0 Eelec + 1.0 Edesol + 0.1 Eair - 0.01 BSA
HADDOCKscore-water = 1.0 Evdw + 0.1 Eelec + 1.0 Edesol + 0.1 Eair
This differs from the defaults setting (defined for protein-protein complexes). We recommend to change two weights for protein-ligand docking:
- Step 8: Apply some ligand-specific setting. For this unfold the Advanced sampling parameter menu:
initial temperature for second TAD cooling step with flexible side-chain at the inferface -> 500
initial temperature for third TAD cooling step with fully flexible interface -> 300
number of MD steps for rigid body high temperature TAD -> 0
number of MD steps during first rigid body cooling stage -> 0
- Step 8: You are ready to submit! Enter your username and password (or the course credentials provided to you). Remember that for this interface you do need guru access.
Again, pre-calculated runs are provided if you have executed the download-run-data.csh and/or download-run-data-full.csh scripts provided in the runs directory (see Setup section).
Analysis of the targeted docking results
Once your run has completed you will be presented with a result page showing the cluster statistics and some graphical representation of the data. Instead, you can also use the precalculated run. Simply unpack the archive, go into the directory and open with your favorite web browser the index.html file to view the results page.
How many clusters have been generated? What is the score difference between the various clusters? Is the top one significantly better in score than the next one?
You can also compare the orientation of the ligand in our models with the orientation of the same ligand in the crystal structure with rifampicin bound in chain C (remember that chain C of that structure corresponds to chain A in the nomemclature of Sennhauser et al.) (3AOD), corresponding to a channel slightly narrower than for chain A (our current chain B in Sennhauser). Or simply use the renumbered 3AOD structure provided in the pdbs directory called 3AOD-renumbered-BCA.pdb to compare the structures in Pymol. In this renumbered structure, we changed the chain IDs such as that the chain binding rifampicin corresponds to chain B of 2J8S which we targeted.
pymol cluster*_1.pdb $WDIR/../pdbs/3AOD-renumbered-BCA.pdb
Note: You should realize that the crystal structure has a limited resolution (3.3Å) and its quality is also limited (see the “Experiments and Validation” page provided by the PDBe for this structure). In general for modelling purposes, it might also be worth considering the recalculated structure from PDB_REDO, the database of updated and optimized X-ray structure models.
Congratulations!
You have completed this tutorial. If you have any questions or suggestions, feel free to contact us via email or by submitting an issue in the appropriate Github repository or asking a question through our support center.