COVID-19 related research projects from the CSB group
- Screening of approved drugs against the SARS-CoV-2 protease
- Screening of approved drugs against the RNA dependent RNA polymerase
- Screening of approved drugs against the ACE2 receptor
- Screening for frequent hitters
- Protocol
The novel coronavirus (SARS-CoV-2) that has emerged from Wuhan, China in December 2019 has spread to almost all countries in the World causing a dramatic number of deaths. The current absence of antiviral treatment against the SARS-CoV-2 urges the scientific community to accelerate the drug discovery research process.
One way to identify potential treatments and to be able to administer it swiftly is to focus on drug repurposing studies, i.e. to investigate the SARS-CoV-2 antiviral potential of drugs that have already been approved for human use.
Screening of approved drugs against the SARS-CoV-2 protease
Proteins that are crucial for the survival and replication of the virus are the most attractive targets for such studies. Here we have focused on the SARS-CoV-2 main protease (3CLpro) that plays an essential role in the virus replication process by screening ~2000 approved drugs (and 6 experimental ones - these can be identified by searching the tables using the term ‘Investigational’) against this particular protein. We used for that HADDOCK2.4, following a pharmacophore-based and a shape-based strategy adapted from our successful participation to the D3R Grand Challanges and described in the following paper:
- P.I. Koukos, L.C. Xue and A.M.J.J. Bonvin. Protein-ligand pose and affinity prediction. Lessons from D3R Grand Challenge 3. J. Comp. Aid. Mol. Des. 33, 83-91 (2019).
More details of the protocol are provided below.
The results of our effort can be seen in the tables and graphs below. The top part presents the results based on top cluster rankings, while the bottom part is based on single structure rankings. The scores (in arbitrary units) correspond to the HADDOCK score calculated as:
HADDOCKscore = 1.0 Evdw + 0.1 Eelec + 1.0 Edesol
where:
- Evdw is the van der Waals intermolecular energy
- Eelec is the electrostatic intermolecular energy
- Edesol is an empirical desolvation energy term.
The plots show one data point per compound. In total there are 2020 compounds, grouped into one of four categories ‘Protease Inhibitors’, ‘Antivirals’, ‘Antiinfectives’ and ‘Other’. The first is made up compounds that are known to inhibit proteases, the second antiviral medications, the third general antiinfectives and the last everything else. The ‘NA’ group corresponds to compounds that have no specific associations - these tend to be things like dietary supplements, aminoacid residues, etc… These are not shown in the plots but are listed in the tables.
The compounds were obtained from Drugbank and pre-processed using the OpenEye Omega software (Hawkins et al. J. Chem. Inf. Model. 50 572-584 (2010)). In addition to these compounds we also obtained some of their active metaqbolites from PubChem. These can be recognised by the prefix CID. For the protease we used PDB entry 6Y2F (Zhang et al. Science, 2020).
Relevance
In total, 7 molecules of the HADDOCK Top 100 drugs (one ranked #2) are under clinical trials in Europe as reported in the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) https://www.clinicaltrialsregister.eu/ctr-search/search?query=covid-19:
- 1 kinase targeting antitumor drug: Imatinib
- 2 antiviral drugs: Lopinavir, Remdesivir
- 3 antiinfective drugs: Azithromycin, ceftriaxone, erythromycin
- 1 angiotensin II receptor antagonist: Telmisartan
Acknowledgments
Our group efforts have been led in major part by Dr. Panagiotis Koukos and Dr. Manon Réau. Dr. Ed Moret from the Department of Pharmaceutical Sciences at Utrecht University has provided valuable expertise on the compounds and their relevance.
Screening results
The plots are zoomable and clickable and groups can be hidden by clicking on them in the legend. Hovering over points in the plot will reveal the name of the compounds.
The tables are sorted based on the HADDOCK score. They are also dynamic, sortable and searchable.
Further experimental screening of promising compounds
We reached out to collaborators in the CARE-IMI consortium to have our most promising compounds experimentally tested. The experiments yielded several compounds with activity against the protease in the micromolar range but subsequent testing revealed them to also be cytotoxic, rendering them less relevant for clinical applications.
Two of the most promising compounds were imatinib and lapatinib, listed as #2 and #5, respectively in our cluster-based ranking.
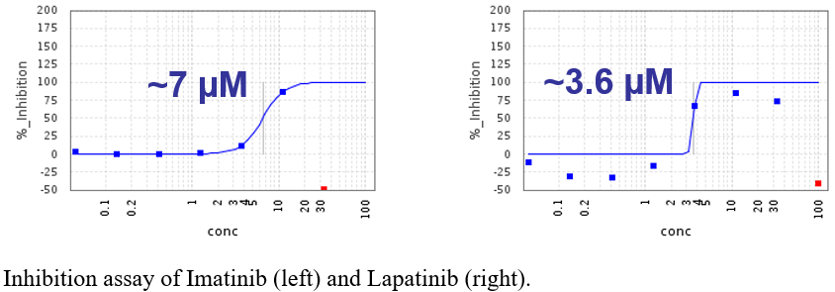
The infection assays, affinity measurements and toxicity assays were carried out in the groups of Frank Kuppeveld at Utrecht University and Johan Neyts at KU Leuven.
Screening of approved drugs against the RNA dependent RNA polymerase
In addition the screening against the main protease we docked the virtual libray against the RNA dependent RNA polymerase (RdRp) as well. We used a truncated from of PDB entry 7BV2 for the docking and the same compound conformers as for the Mpro screening.
Screening results
Screening of approved drugs against the ACE2 receptor
In addition the screening against the main protease we docked the virtual libray against the RNA dependent RNA polymerase (RdRp) as well. We used a truncated from of PDB entry 1R4L for the docking and the same compound conformers as for the Mpro screening.
Screening results
Screening for frequent hitters
As can be seen in the results above Ceftolozane is the top-scoring compound for two of the three
screens we have performed so far. This prompted us to dock the top-scoring compounds against an
unrelated target - Dihydrofolate reductase (DHFR). Taking the top 20 compounds from the three screens
and excluding the overlapping targets gave us a list of 48 compounds. The results of the screen can be
seen below. It is clear that some of our top scoring compounds can be classified as frequent hitters
as they seem to “stick” to multiple targets, perhaps owing to their large size and/or polar nature.
See the protocol section for details regarding the scores reported for DHFR.
Screening results
Protocol
Virtual library
We considered approved drugs from the DrugBank dataset with molecular weight < 750 g/mol and > 5 heavy atoms. Some of these drugs act as prodrugs, i.e. they are inactive compounds that are metabolized into their active form after administration. When the data was available we collected the active form of the documented prodrugs, referred here as « Active Metabolites », from the PubChem database. We ended up with 1977 drugs and 50 active metabolites and we generated up to 500 conformers per compound using OpenEye.
Shape and pharmacophore-based protocols
Template compounds
The rational behind HADDOCK docking is to make use of experimental information to guide the docking. Herein, we took advantage of the large amount of high quality holo structures of the SARS-CoV-2 3CLpro and related proteins (> 90% identity) published in the Protein Data Bank. Among those crystallographic data, 66 molecules are non-covalent and covalent active-site fragments from the large XChem crystallographic fragment screen against 3CLpro performed by Diamond. In total, we collected 92 molecules targeting the 3CLpro(-related) binding site.
Shape based docking - HADDOCK
We identified one crystallographic template to be used for the docking of every target compound in the virtual library. For the selection, we calculated the Tanimoto coefficient computed over the Maximum Common Substructure (MCS) between target and template compounds and selected the template with the highest Tanimoto coefficient. After superimposing all templates we transformed the heavy atoms of their compounds into shape beads and defined restraints between the beads and heavy atoms of the target compound to guide it in the binding pocket.
Pharmacophore based docking - HADDOCK
Similarly, each coumpound in the virtual library was associated to the most similar crystallographic template in terms of 2D pharmacophore description. To do so, we calculated the pairwise Tanimoto coefficient between the 2D pharmacophore fingerprints of the compounds from the virtual library and the template compounds. The binding information of the template compound was then used to build a shape in the 3CLpro binding site consisting of one bead per atom, each bead being associated to a pharmacophore feature (or no feature) as computed with RDKIT. Docking restraints were imposed to orient the pharmacophore features of the drugs and active metabolites towards the corresponding feature of their associated shape.
Distance restraint-based protocol
RdRp screening
We used a protocol based on the definition of distance restraints for the screenign of the virtual library against RdRp. The definition of the binding site was extracted from pdb entry 7BV2, after calculating atomic contacts between the bound compound (remdesivir monophosphate) and the protein, using a distance cutoff of 5A. None of the restraints were discarded during the simulation. We lowered the scaling constant of the intermolecular energies to a thousandth of their original value to allow the compounds to more effectively penetrate into the buried binding pocket. Sampling was the same as for the main protease. We only took intermolecular vdW and electrostatics energies and desolvation potential into account for ranking the compounds.
ACE2 screening
The protocol we followed was identical to the one described above (see RdRp screening) with the exception of using pdb entry 1R4L as the receptor template.
DHFR screening
We used the distance-based protocol for DHFR screening (screening for frequent hitters) as well. We used PDB entry 6NND. The screen was repeated three times to test the reproducibility of the scores obtained by the protocol. With the exception of a handful of compounds with high deviation between runs there was excellent agreement between replicas.