

HADDOCK basic protein-protein docking tutorial
This tutorial consists of the following sections:
- Introduction
- Setup
- HADDOCK general concepts
- Inspecting and preparing E2A for docking
- Inspecting and preparing HPR for docking
- Adding a phosphate group
- Setting up the docking run
- Analysing the results
- Biological insights
- Comparison with the reference structure
- Additional docking runs
Introduction
This tutorial will demonstrate the use of HADDOCK for predicting the structure of a protein-protein complex from NMR chemical shift perturbation (CSP) data. Namely, we will dock two E. coli proteins involved in glucose transport: the glucose-specific enzyme IIA (E2A) and the histidine-containing phosphocarrier protein (HPr). The structures in the free form have been determined using X-ray crystallography (E2A) (PDB ID 1F3G) and NMR spectroscopy (HPr) (PDB ID 1HDN). The structure of the native complex has also been determined with NMR (PDB ID 1GGR). These NMR experiments have also provided us with an array of data on the interaction itself (chemical shift perturbations, intermolecular NOEs, residual dipolar couplings, and simulated diffusion anisotropy data), which will be useful for the docking. For this tutorial, we will only make use of inteface residues identified from NMR chemical shift perturbation data as described in Wang et al, EMBO J (2000).
For this tutorial we will make use of the HADDOCK2.2 webserver. A description of our web server can be found in the following publications:
-
G.C.P van Zundert, J.P.G.L.M. Rodrigues, M. Trellet, C. Schmitz, P.L. Kastritis, E. Karaca, A.S.J. Melquiond, M. van Dijk, S.J. de Vries and A.M.J.J. Bonvin. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol., 428, 720-725 (2015).
-
S.J. de Vries, M. van Dijk and A.M.J.J. Bonvin. The HADDOCK web server for data-driven biomolecular docking. Nature Protocols, 5, 883-897 (2010). Download an open version here.
Throughout the tutorial, coloured text will be used to refer to questions or instructions, and/or PyMOL commands.
This is a question prompt: try answering it! This an instruction prompt: follow it! This is a PyMOL prompt: write this in the PyMOL command line prompt!
Setup
In order to run this tutorial you will need to have the following software installed: PyMOL.
Also, if not provided with special workshop credentials to use the HADDOCK portal, make sure to register in order to be able to submit jobs. Use for this the following registration page: https://alcazar.science.uu.nl/services/HADDOCK2.2/signup.html.
HADDOCK general concepts
HADDOCK (see https://www.bonvinlab.org/software/haddock2.2) is a collection of python scripts derived from ARIA (https://aria.pasteur.fr) that harness the power of CNS (Crystallography and NMR System – https://cns-online.org) for structure calculation of molecular complexes. What distinguishes HADDOCK from other docking software is its ability, inherited from CNS, to incorporate experimental data as restraints and use these to guide the docking process alongside traditional energetics and shape complementarity. Moreover, the intimate coupling with CNS endows HADDOCK with the ability to actually produce models of sufficient quality to be archived in the Protein Data Bank.
A central aspect to HADDOCK is the definition of Ambiguous Interaction Restraints or AIRs. These allow the translation of raw data such as NMR chemical shift perturbation or mutagenesis experiments into distance restraints that are incorporated in the energy function used in the calculations. AIRs are defined through a list of residues that fall under two categories: active and passive. Generally, active residues are those of central importance for the interaction, such as residues whose knockouts abolish the interaction or those where the chemical shift perturbation is higher. Throughout the simulation, these active residues are restrained to be part of the interface, if possible, otherwise incurring in a scoring penalty. Passive residues are those that contribute for the interaction, but are deemed of less importance. If such a residue does not belong in the interface there is no scoring penalty. Hence, a careful selection of which residues are active and which are passive is critical for the success of the docking.
The docking protocol of HADDOCK was designed so that the molecules experience varying degrees of flexibility and different chemical environments, and it can be divided in three different stages, each with a defined goal and characteristics:
-
1. Randomization of orientations and rigid-body minimization (it0)
In this initial stage, the interacting partners are treated as rigid bodies, meaning that all geometrical parameters such as bonds lengths, bond angles, and dihedral angles are frozen. The partners are separated in space and rotated randomly about their centres of mass. This is followed by a rigid body energy minimization step, where the partners are allowed to rotate and translate to optimize the interaction. The role of AIRs in this stage is of particular importance. Since they are included in the energy function being minimized, the resulting complexes will be biased towards them. For example, defining a very strict set of AIRs leads to a very narrow sampling of the conformational space, meaning that the generated poses will be very similar. Conversely, very sparse restraints (e.g. the entire surface of a partner) will result in very different solutions, displaying greater variability in the region of binding. -
2. Semi-flexible simulated annealing in torsion angle space (it1)
The second stage of the docking protocol introduces flexibility to the interacting partners through a three-step molecular dynamics-based refinement in order to optimize interface packing. It is worth noting that flexibility in torsion angle space means that bond lengths and angles are still frozen. The interacting partners are first kept rigid and only their orientations are optimized. Flexibility is then introduced in the interface, which is automatically defined based on an analysis of intermolecular contacts within a 5Å cut-off. This allows different binding poses coming from it0 to have different flexible regions defined. Residues belonging to this interface region are then allowed to move their side-chains in a second refinement step. Finally, both backbone and side-chains of the flexible interface are granted freedom. The AIRs again play an important role at this stage since they might drive conformational changes. -
3. Refinement in Cartesian space with explicit solvent (water)
The final stage of the docking protocol immerses the complex in a solvent shell so as to improve the energetics of the interaction. HADDOCK currently supports water (TIP3P model) and DMSO environments. The latter can be used as a membrane mimic. In this short explicit solvent refinement the models are subjected to a short molecular dynamics simulation at 300K, with position restraints on the non-interface heavy atoms. These restraints are later relaxed to allow all side chains to be optimized.
The performance of this protocol of course depends on the number of models generated at each step. Few models are less probable to capture the correct binding pose, while an exaggerated number will become computationally unreasonable. The standard HADDOCK protocol generates 1000 models in the rigid body minimization stage, and then refines the best 200 – regarding the energy function - in both it1 and water. Note, however, that while 1000 models are generated by default in it0, they are the result of five minimization trials and for each of these the 180º symmetrical solution is also sampled. Effectively, the 1000 models written to disk are thus the results of the sampling of 10.000 docking solutions.
The final models are automatically clustered based on a specific similarity measure - either the positional interface ligand RMSD (iL-RMSD) that captures conformational changes about the interface by fitting on the interface of the receptor (the first molecule) and calculating the RMSDs on the interface of the smaller partner, or the fraction of common contacts (current default) that measures the similarity of the intermolecular contacts. For RMSD clustering, the interface used in the calculation is automatically defined based on an analysis of all contacts made in all models.
Inspecting and preparing E2A for docking
We will now inspect the E2A structure. For this start PyMOL and in the command line window of PyMOL (indicated by PyMOL>) type:
fetch 1F3G
show cartoon
hide lines
show sticks, resn HIS
You should see a backbone representation of the protein with only the histidine side-chains visible. Try to locate the histidines in this structure.
Is there any phosphate group present in this structure?
Note that you can zoom on the histidines by typing in PyMOL:
Revert to a full view with:
As a preparation step before docking, it is advised to remove any irrelevant water and other small molecules (e.g. small molecules from the crystallisation buffer), however do leave relevant co-factors if present. For E2A, the PDB file only contains water molecules. You can remove those in PyMOL by typing:
Now let’s vizualize the residues affected by binding as identified by NMR. From Wang et al, EMBO J (2000) the following residues of E2A were identified has having significant chemical shift perturbations:
38,40,45,46,69,71,78,80,94,96,141
We will now switch to a surface representation of the molecule and highlight the NMR-defined interface. In PyMOL type the following commands:
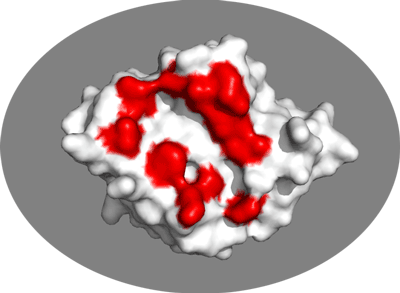
Inspect the surface.
Do the identified residues form a well defined patch on the surface? Do they form a contiguous surface?
The answer to the last question should be no: We can observe residue in the center of the patch that do not seem significantly affected while still being in the middle of the defined interface. This is the reason why in HADDOCK we also define “passive” residues that correspond to surface neighbors of active residues. These can be selected manually, or more conveniently you can let the HADDOCK server do it for you (see Setting up the docking run below).
As final step save the molecule as a new PDB file which we will call: e2a_1F3G.pdb
For this in the PyMOL menu on top select:
File -> Save molecule… Select 1F3G and click on the save button Name your file e2a_1F3G.pdb and note its location
Inspecting and preparing HPR for docking
We will now inspect the HPR structure. For this start PyMOL and in the command line window of PyMOL type:
fetch 1HDN
show cartoon
hide lines
Since this is an NMR structure it does not contain any water molecules and we don’t need to remove them.
Let’s vizualize the residues affected by binding as identified by NMR. From Wang et al, EMBO J (2000) the following residues were identified has having significant chemical shift perturbations:
We will now switch to a surface representation of the molecule and highlight the NMR-defined interface. In PyMOL type the following commands:
Again, inspect the surface.
Do the identified residues form a well defined patch on the surface? Do they form a contiguous surface?
Now since this is an NMR structure, it actually consists of an ensemble of models. HADDOCK can handle such ensemble, using each conformer in turn as starting point for the docking. We however recommend to limit the number of conformers used for docking, since the number of conformer combinations of the input molecules might explode (e.g. 10 conformers each will give 100 starting combinations and if we generate 1000 ridig body models (see HADDOCK general concepts above) each combination will only be sampled 10 times).
Now let’s vizualise this NMR ensemble. In PyMOL type:
hide all
show ribbon
set all_states, on
You should now be seing the 30 conformers present in this NMR structure. To illustrate the potential benefit of using an ensemble of conformations as starting point for docking let’s look at the side-chains of the active residues:
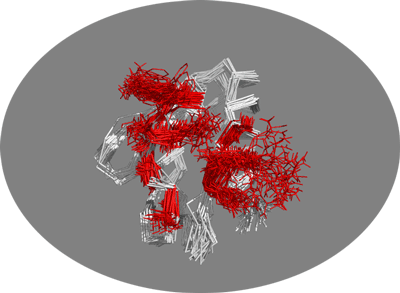
You should be able to see the amount of conformational space sampled by those surface side-chains. You can clearly see that some residues do sample a large variety of conformations, one of which might lead to much better docking results.
Note: Pre-sampling of possible conformational changes can thus be beneficial for the docking, but again do limit the number of conformers used for the docking (or increase the number of sampled models, which is possible in the expert interface of the HADDOCK portal - the default access level is however only easy - for a higher level access do request it via email).
As final step, save the molecule as a new PDB file which we will call: hpr-ensemble.pdb For this in the PyMOL menu select:
File -> Save molecule… Select 1HDN and click on the save button Name your file hpr-ensemble.pdb and note its location
Adding a phosphate group
Since the biological function of this complex is to transfer a phosphate group from one protein to another, via histidines side-chains, it is relevant to make sure that a phosphate group be present for docking. As we have seen above none is currently present in the PDB files. HADDOCK does support a list of modified amino acids which you can find at the following link: https://alcazar.science.uu.nl/services/HADDOCK2.2/library.html.
Check the list of supported modified amino acids. What is the proper residue name for a phospho-histidine in HADDOCK?
In order to use a modified amino-acid in HADDOCK, the only thing you will need to do is to edit the PDB file and change the residue name of the amino-acid you want to modify. Don’t bother deleting irrelevant atoms or adding missing ones, HADDOCK will take care of that. For E2A, the histidine that is phosphorylated has residue number 90. In order to change it to a phosphorylated histidine do the following:
Edit the PDB file (e2a_1F3G.pdb) in your favorite editor Change the name of histidine 90 to NEP Save the file (as simple text file) under a new name, e.g. e2aP_1F3G.pdb
Note: The same procedure can be used to introduce a mutation in an input protein structure.
Setting up the docking run
We will now launch the docking run. For this we will make us of the easy interface of the HADDOCK web server:
https://alcazar.science.uu.nl/services/HADDOCK2.2/haddockserver-easy.html
Note: The blue bars on the server can be folded/unfolded by clicking on the arrow on the right
-
Step1: Define a name for your docking run, e.g. E2A-HPR.
-
Step2: Input the first protein PDB file. For this unfold the First Molecule menu.
First molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (for this particular case) PDB structure to submit -> Browse and select e2aP_1F3G.pdb (the file you edited to modify the histidine) Active residues (directly involved in the interaction) -> 38,40,45,46,69,71,78,80,94,96,141 Define passive residues automatically around the active residues -> check box
- Step3: Input the proteins PDB files. For this unfold the Second Molecule menu.
First molecule: where is the structure provided? -> “I am submitting it” Which chain to be used? -> All (for this particular case) PDB structure to submit -> Browse and select hpr-ensemble.pdb (the file you saved) Active residues (directly involved in the interaction) -> 15,16,17,20,48,49,51,52,54,56 Define passive residues automatically around the active residues -> check box
- Step 4: You are ready to submit!
Enter your username and password (or the course credentials provided to you).
Upon submission you will first be presented with a web page containing a link to the results page, but also an importantly a link to a haddockparameter file (simple text format) containing all settings and input data of your run.
We strongly recommend to save this haddockparameter file since it will allow you to repeat the run by simple upload into the file upload inteface of the HADDOCK webserver. It can serve as input reference for the run. This file can also be edited to change a few parameters for examples. An excerpt of this file is shown here:
HaddockRunParameters ( runname = 'E2A-HPR', auto_passive_radius = 6.5, create_narestraints = True, delenph = True, ranair = False, cmrest = False, kcont = 1.0, surfrest = False, ksurf = 1.0, noecv = True, ncvpart = 2.0, structures_0 = 1000, ntrials = 5, ...
Click now on the link to the results page. While your input data are being validated and processed the page will show:

During this stage the PDB and eventually provided restraint files are being validated. Further the server makes use of Molprobity to check side-chain conformations, eventually swap them (e.g. for asparagines) and define the protonation state of histidine residues. Once this has been successfully done, the page will indicated that your job is first QUEUED, and then RUNNING.

The page will automatically refresh and the results will appear upon completions (which can take between 1/2 hour to several hours depending on the size of your system and the load of the server). You will be notified by email once your job has successfully completed.
Analysing the results
Once your run has completed you will be presented with a result page showing the cluster statistics and some graphical representation of the data (and if registered, you will also be notified by email). Such an example output page can be found here in case you don’t want to wait for the results of your docking run.
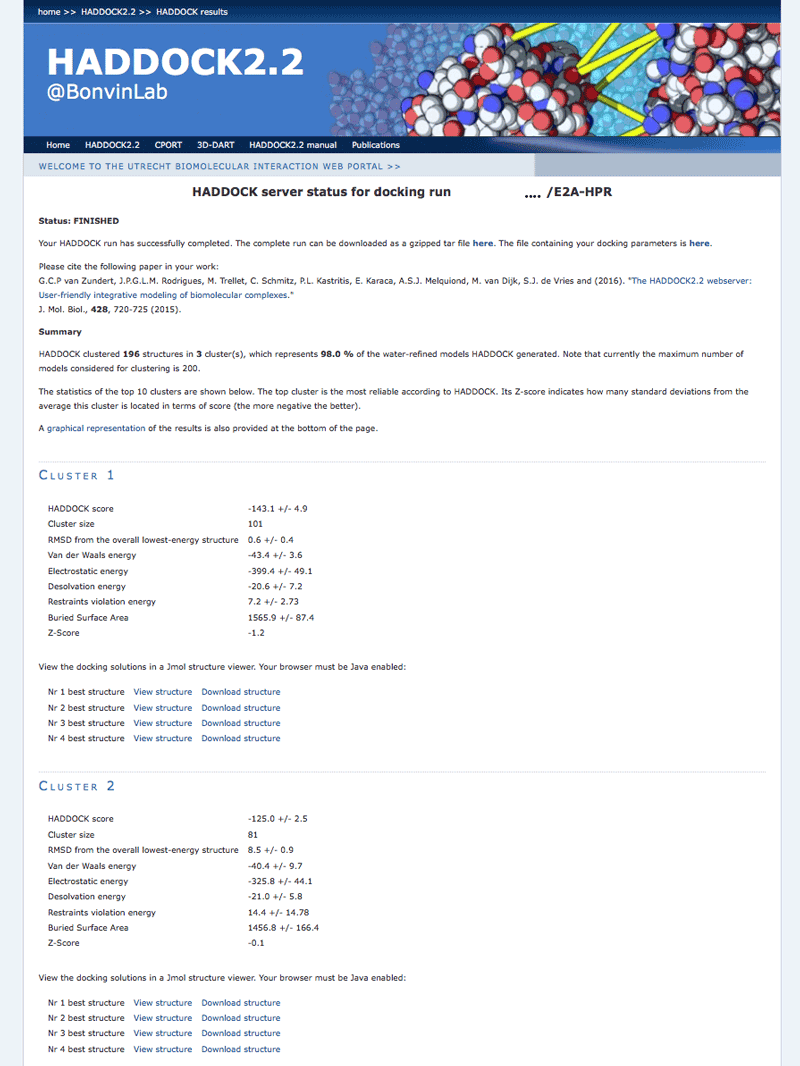
Inspect the result page How many clusters are generated?
Note: The bottom of the page gives you some graphical representations of the results, showing the distribution of the solutions for various measures (HADDOCK score, van der Waals energy, …) as a function of the RMSD from the best generated model (the best scoring model).
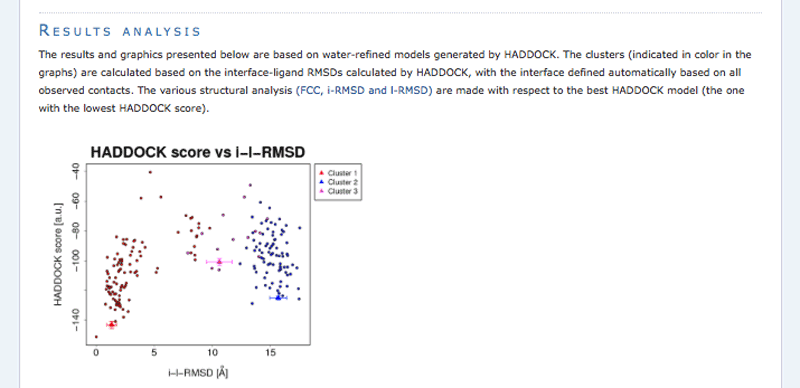
The ranking of the clusters is based on the average score of the top 4 members of each cluster. The score is calculated as:
HADDOCKscore = 1.0 * Evdw + 0.2 * Eelec + 1.0 * Edesol + 0.1 * Eair
where Evdw is the intermolecular van der Waals energy, Eelec the intermolecular electrostatic energy, Edesol represents an empirical desolvation energy term adapted from Fernandez-Recio et al. J. Mol. Biol. 2004, and Eair the AIR energy. The cluster numbering reflects the size of the cluster, with cluster 1 being the most populated cluster. The various components of the HADDOCK score are also reported for each cluster on the results web page.
Consider the cluster scores and their standard deviations. Is the top ranked cluster significantly better than the second one? (This is also reflected in the z-score).
In case the scores of various clusters are within standard devatiation from each other, all should be considered as a valid solution for the docking. Ideally, some additional independent experimental information should be available to decide on the best solution. In this case we do have such a piece of information: the phosphate transfer mechanism (see Biological insights below).
Let’s now visualise the various solutions.
Download and save to disk the first model of each cluster
Then start PyMOL and load each cluster representative:
File menu -> Open -> select cluster1_1.pdb
Repeat this for each cluster. Once all files have been loaded, type in the PyMOL command window:
show cartoon
util.cbc
hide lines
Let’s then superimpose all models on chain A of the first cluster:
select cluster1_1 and chain A
align cluster2_1, sele
Repeat the align command for each cluster representative.
This will align all clusters on chain A (E2A), maximizing the differences in the orientation of chain B (HPR).
Examine the various clusters. How does the orientation of HPR differ between them?
Note: You can turn on and off a cluster by clicking on its name in the left panel of the PyMOL window.
Let’s now check if the active residues which we defined are actually part of the interface. In the PyMOL command window type:
Are the active residues in the interface?
Biological insight
The E2A-HPR complex is involved in phosphate-transfer, in which a phosphate group attached to histidine 90 of E2A (which we named NEP) is transferred to a histidine of HPR. As such, the docking models should make sense according to this information, meaning that two histidines should be in close proximity at the interface. Using PyMOL, check the various cluster representatives (we are assuming here you have performed all PyMOL commands of the previous section):
select histidines, resn HIS+NEP
show spheres, histidines
util.cnc
First of all, has the phosphate group been poperly generated?
Note: You can zoom on the phosphorylated histidine using the following PyMOL command:
Zoom back to all visible molecules with
Now inspect each cluster in turn and check if histidine 90 of E2A is in close proximity to another histidine of HPR. To facilitate this analysis, view each cluster in turn (use the left panel to activate/desactivate the various clusters by clicking on their name).
Based on this analysis, which cluster does satisfy best the biolocal information?
Is this cluster also the best ranked one?
Comparison with the reference structure
As explained in the introduction, the structure of the native complex has been determined by NMR (PDB ID 1GGR) using a combination of intermolecular NOEs and dipolar coupling restraints. We will now compare the docking models with this structure.
If you still have all cluster representative open in PyMOL you can proceed with the sub-sequent analysis, otherwise load again each cluster representative as described above. Then, fetch the reference complex by typing in PyMOL:
fetch 1GGR
show cartoon
color yellow, 1GGR and chain A
color orange, 1GGR and chain B
The number of chain B in this structure is however different from the HPR numbering in the structure we used: It starts at 301 while in our models chain B starts at 1. We can change the residue numbering easily in PyMol with the following command:
alter (chain B and 1GGR), resv -=300
Then superimpose all cluster representatives on the reference structure, using the entire chain A (E2A):
select 1GGR and chain A
align cluster1_1, sele, cycles=0
Repeat the align command for each cluster representative.
Does any of the cluster representatives ressemble the reference NMR structure? In case you found a reasonable prediction, what is its cluster rank?
In the blind protein-protein prediction experiment CAPRI (Critical PRediction of Interactions), a measure of the quality of a model is the so-called ligand-RMSD (l-RMSD). It is calculated by fitting on the receptor chain (E2A or chain A in our case) and calculating the RMSD on the backbone of the ligand (HPR or chain B in our case). This can be done in PyMOL with the following command:
rms_cur cluster1_1 and chain B, 1GGR
In CAPRI, the l-RMSD value defines the quality of a model:
- acceptable model: l-RMSD<10Å
- medium quality model: l-RMSD<5Å
- high quality model: l-RMSD<1Å
What is based on this CAPRI criterion the quality of the best model?
Congratulations!
You have completed this tutorial. If you have any questions or suggestions, feel free to contact us via email or asking a question through our support center.
Additional docking runs
If you are curious and want learn more about HADDOCK and the impact of the input data on the docking results, consider performing and analysing, as described above, the following runs:
- Same run as above, but without defining the phosphorylated histidine
- Same run as above, but using only the first model of the HPR ensemble (edit the file to extract it)
And check also our education web page where you will find more tutorials!