

Homology Modeling of the mouse MDM2 protein
General Overview
This tutorial is divided in 5 sections, each representing (roughly) a step of the homology modeling procedure:
- A bite of theory
- Using Uniprot to retrieve sequence information
- Finding homologues of known structure using HMMER
- Choosing a template from the list of homologues
- Modeling mouse MDM2 using MODELLER
A bite of theory
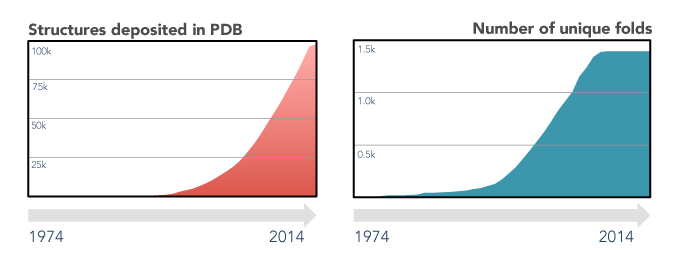
The last decades of scientific advances in the fields of protein biology revealed the extent of both the protein sequence and structure universes. Protein sequences databases currently hold tens of millions of entries (source) and are foreseen to continue growing exponentially, driven by high-throughput sequencing efforts. On the other hand, the number of experimental protein structures is two orders of magnitude smaller (source), and that of unique folds has remained virtually unchanged since 2008. This apparent stagnation of the protein structure universe is a boon for structure prediction enthusiasts, as finding a sequence without a structurally characterized close homologue is, nowadays, quite rare.
There are many methods for predicting the three-dimensional structure of proteins from their sequence, most of which fall in one of three broad categories. Of this triumvirate, homology modeling is the most reliable class of methods, with an estimated accuracy close to a low-resolution experimental structure (source). The two others, molecular threading and ab initio modeling, are usually of interest only if homology modeling is not an option.
Homology modeling is then a structure prediction method - worth noting, not exclusively for proteins - that exploits the robustness of protein structure to changes in primary sequence. When protein crystallography became routine in the 1980s, researchers started analyzing and comparing high-resolution structures. In doing so, they quickly realized that evolutionarily related proteins shared common structural features and that the extent of this structural similarity directly correlated with the sequence similarity (source). To maintain structure and function, certain amino acids in the protein sequence suffer a stronger selective pressure, evolving either slower than expected or within specific constraints, such as chemical similarity. Combining these and other observations, early computational structural biologists created the first homology modeling algorithms in the late 1980s/early 1990s.
Using Uniprot to retrieve sequence information
Your goal is to create a model of the MDM2 mouse protein, in particular of its N-terminal region that binds the p53 trans-activation domain. So, where to start?
The Uniprot database is an online resource offering access to all known protein sequences. Besides raw sequence data, Uniprot aggregates information from several other databases such as the The Worldwide PDB (wwPDB) that archives information about the 3D structures of proteins, nucleic acids, and complex assemblies and ensures that the PDB is freely and publicly available to the global community, NCBI Pubmed, KEGG, Pfam, and many others. The wwPDB itself consists of several sites that all provide access in their own way to the wwPBD core database together with various associated services: The Research Collaboratory for Structural Bioinformatics PDB (RCSB), PDB Europe (PDBe) and PDB Japan (PDBj), together with the Biological Magnetic Resonance Data Bank (BMRB) that collects NMR data. These features of Uniprot makes it an obvious go-to resource when looking for information on any protein. There are two collections of sequences: Swiss-Prot, whose entries undergo manual annotation and revision, and TrEMBL, where the annotation is unsupervised. Consequently, if the entry for a particular protein of interest belongs to Swiss-Prot, it will be marked by a golden star/icon meaning its contents are very likely (but not blindly!) reliable.
Find the mouse MDM2 entry in Uniprot using the search box on the home page.
Take the time to browse through the Uniprot page of mouse MDM2. The header of the page lists the protein, gene, and organism names for this particular entry, as well as its unique Uniprot accession code. On the left, below the header, there is a sidebar listing the several sections of the page. You can use these to navigate directly to the ‘Structure’ section to verify if there are already published experimental structures for mouse MDM2. Fortunately, there aren’t any; otherwise this tutorial would end here.
Besides reporting on experimental structures, Uniprot links to portals such as the SWISS-MODEL Repository, and ModBase, which regularly cross-reference sequence and structure databases in order to build homology models. These automated protocols are configured to create models only under certain conditions, such as sufficient sequence identity and coverage. Still, the template identification, target/template alignment, and modeling options are unsupervised, which may lead to severe errors in some cases. In general, these models offer a quick peek of what fold(s) a particular sequence can adapt and may as well serve as a starting point for further refinement and analyses. Nevertheless, if the model will be a central part of a larger study, it might be worth to invest time and effort in modeling a particular protein of interest with a set of dedicated protocols.
The following tab, “Family & Domains”, lists structural and domain information derived either from experiments or by similarity to other entries. For the mouse MDM2 protein, it shows that it contains a SWIB domain and two zinc fingers and that it interacts with proteins such as USP2, PYHIN1, RFFL, RNF34, among others. Additional information displayed in the text offers additional insights on binding partners and interfaces.
Which region(s) of MDM2 bind p53 and which of those bind to the trans-activation domain?
From the introduction, you know that our region of interest in MDM2 interacts with the trans-activation region of p53 and does not ubiquitinate it. The small print under the “Domain” header gives clues regarding possible p53 interfaces: “Region I is sufficient for binding p53”; “the RING finger domain […] is also essential for [MDM2] ubiquitin ligase E3 activity toward p53”. It seems, therefore, that Region I is our modeling target, but besides this annotation, it is not listed anywhere on the Uniprot page. While this mystery has plenty of possible solutions, the easiest of which would be to search for a publication on the MDM2 domain organisation, keep to the Uniprot page to find an answer.
Browsing further down the page, the “Sequences” tab lists the several isoforms of this particular protein as they have been observed. One of these is classified as “canonical” while others are products of splicing events or mutations. The notes on isoform MDM2-p76 reveal that it lacks the first 49 amino acids and that it does not bind p53. The interaction occurs then through the N-terminal of MDM2. Linking this information with that of the domain organization hints that the first region (positions 1-110) is very likely our modeling target. This selection can be further refined by choosing only the region comprising the SWIB domain (positions 27-107). Choose either the first region (positions 1-110), the SWIB domain, or whatever seems best in your opinion.
Why can the first ~20 amino acids of MDM2 be neglected for the modeling?
Clicking on the “position(s)” column of a particular region/domain opens a new window showing the corresponding sequence as well as the region in the context of the full sequence. Although this window provides a shortcut to launch a BLAST similarity search against the UniprotKB (or another) database, there are other more sensitive methods for this purpose. For now, pay attention to the sequence and its format. Named FASTA after the software program it was first implemented in, it is perhaps the most widely used file format in bioinformatics, owing surely to its readability for both humans and machines.
>sp|P23804|1-110
MCNTNMSVSTEGAASTSQIPASEQETLVRPKPLLLKLLKSVGAQNDTYTMKEIIFYIGQY
IMTKRLYDEKQQHIVYCSNDLLGDVFGVPSFSVKEHRKIYAMIYRNLVAVFor each sequence in the file, it contains a header line starting with > followed by an
identifier. In the Uniprot page, the identifier contains the entry’s collection (sp - Swiss-Prot),
accession code, and region of the sequence. The information on this header is used by several
programs in many different ways, so it makes sense to keep it simple and readable.
Change the identifier to something more meaningful and human readable (e.g. MDM2_MOUSE).
The next line(s) contains the sequence in the standard one-letter code. Any character other than an upper case letter will cause some (not all) programs to throw an error about the format of the sequence. Although there is not a strictly enforced character limit, it is customary to split the sequence into multiple lines of 80 characters each. This limit, as many others based on character length, is a legacy from the old days when screen resolutions were small or terminals the only way of interfacing with the computer. Nevertheless, some programs will complain, or even worse, truncate, lines longer than these 80 characters, so it is wise to respect this limit!
Copy the FASTA-formatted sequence to a text file and save it under an appropriate name (e.g. MDM2_MOUSE.fasta). Save the file in the home directory, Downloads/ folder, or any other easily accessible location.
Now that you have a sequence, the following step is to find a suitable homolog to use in the modeling protocol. The several homology modeling methods available online, such as the HHpred web server, need only this sequence to start the entire procedure. After a few minutes or hours, depending on the protocol, these servers produce models and a set of quality criteria to help the user make a choice. The downside of using a web server is that, usually, the modeling protocol is a ‘black box’. It is impossible to control settings beyond which templates and alignment to use. It is important, however, to understand what is happening behind the scenes, to make conscious choices and grasp the limitations of each method and model. Therefore, this tutorial uses a set of locally installed programs to search for templates, build the models, and evaluate their quality.
Finding homologues of known structure using HMMER
The template is the structurally-resolved homolog that serves as a basis for the modeling. The query, on the other hand, is the sequence being modelled. This standard nomenclature is used by several web servers, software programs, and literature in the field of structure modeling. The first step in any modeling protocol is, therefore, to find a suitable template for the query.
As mentioned before, there are computational methods that perform similarity searches against databases of known sequences. BLAST is the most popular of such methods, and probably the most popular bioinformatics algorithm, with two of its versions in the top 20 of the most cited papers in history (source). It works by finding fragments of the query that are similar to fragments of sequences in a database and then merging them into full alignments (source). Another class of similarity search methods uses the query sequence to seed a general profile sequence that summarises significant features in those sequences, such as the most conserved amino acids. This profile sequence is then used to search the database for homologues. This approach used in PSI-BLAST, an iterative version of BLAST, and also in HMMER, which employs an entirely different statistical framework.
What is the advantage of searching sequence databases with a “profile” sequence?
Whichever the sequence search algorithm, the chances are that, after running through the database, it returns a (hopefully) long list of results. Each entry in this list refers to a particular sequence, the hit, which was deemed similar to the query. It will contain the sequence alignment itself and also some quantitative statistics, namely the sequence similarity, the bit score of the alignment, and its expectation (E) value. Sequence similarity is a quantitative measure of how evolutionarily related two sequences are. It is essentially a comparison of every amino acid to its aligned equivalent. There are three possible outcomes out of this comparison: the amino acids are exactly the same, i.e. identical; they are different but share common physicochemical characteristics, i.e. similar; they are neither. It is also possible that the alignment algorithm introduced gaps in either of the sequences, meaning that there was possibly an insertion or a deletion event during evolution. While identity is straightforward, similarity depends on specific criteria that group amino acids together, e.g. D/E, K/R/H, F/Y/W. The bit score is the likelihood that the query sequence is truly a homologue of the hit, as opposed to a random match. The E-value represents the number of sequences that are expected to have a bit score higher than that of this particular alignment just by chance, given the database size. In essence, a very high bit score and a very small E-value is an assurance that the alignment is indeed significant and that this hit is likely a true homologue of the query sequence.
Our goal is to search for homologues in a sequence database containing exclusively proteins of known structure, such as RCSB PDB. This database is available in text format at the RCSB website and as a selection in most of the homology search web servers. Given the rather small size of these databases (~100k sequences), for reasonably sized sequences, searches take only a few seconds on a laptop.
Start by creating a folder to store all the information related to the modeling process. To keep the files easily accessible, create the folder in the home directory, for example.
cd $HOME
mkdir mdm2_modeling
cd mdm2_modeling
Assuming the Uniprot sequence file is in the Downloads folder, copy it to the newly created folder
and launch a phmmer search against the RCSB PDB sequence database. phmmer is part of the HMMER
suite and searches a query protein sequence against a database. Behind the scenes, it builds a
profile HMM from the query sequence and uses this profile to search the database for homologs
(source).
cp $HOME/Downloads/MDM2_MOUSE.fasta .
phmmer --notextw -o psa.out -A psa.sto MDM2_MOUSE.fasta $MOLMOD_DATA/pdb_seqres.txt
Adapt the file names accordingly!
Take a moment to inspect that command. (Most) GNU/Linux commands start with the command or program
name, after which follow arguments and options. Arguments are usually positional, meaning that
there is a specific order that must be respected. A dash (-) symbol indicates an option. Some
options do not take any values and act as a simple switch while others require a value. Usually, a
well-written program supports a -h or --help option that will print useful information
containing the program description and usage instructions. In this particular example, phmmer is
the program being executed, --notextw, -o psa.out, and -A psa.sto are options, and the
remaining are arguments. The output of phmmer -h shows that -notextw and -o are both
output-related, referring to the maximum line width and output file name respectively. The
remaining option, -A, forces HMMER to write the full alignments to a separate file. As for the
arguments, it shows that the first must be the sequence file and the second the sequence database.
Additionally, there are certain instructions that are not necessarily part of the command or
explained in its documentation. These are part of the GNU/Linux system and deal mostly with the
execution of the command and its input/output. This tutorial will use the ampersand (&) and the
redirection (>) symbols repeatedly. The first runs a program in the background and prevents the
terminal session from being blocked while the second redirects the output of the command to a file.
For more information, refer to a generic tutorial on command-line usage
(example).
Back to the homology modeling, depending on which MDM2 sequence HMMER used to seed the search, the results will vary.
Open and inspect the phmmer output file. leafpad psa.out &
The output file, psa.out, contains detailed information on each hit of the database considered
homologous to the query. The first lines, starting with a # character, show information on the
search parameters and the version of HMMER used to carry it out. This information is always useful;
it allows a user to trace back the specifics of a particular step of the modeling, therefore
allowing some degree of reproducibility.
The interesting bit comes next, after a line stating the name and length of the query sequence, as
read from the input FASTA file.
Query: MDM2_MOUSE [L=110]
Scores for complete sequences (score includes all domains):
--- full sequence --- --- best 1 domain --- -#dom-
E-value score bias E-value score bias exp N Sequence Description
------- ------ ----- ------- ------ ----- ---- -- -------- -----------
1e-64 218.8 0.1 1.2e-64 218.6 0.1 1.0 1 1z1m_A mol:protein length:119 Ubiquitin-protein ligase E3 Mdm2
1.2e-64 218.5 0.1 1.4e-64 218.3 0.1 1.0 1 2lzg_A mol:protein length:125 E3 ubiquitin-protein ligase Mdm2
2.2e-62 211.3 0.1 2.4e-62 211.2 0.1 1.0 1 2mps_A mol:protein length:107 E3 ubiquitin-protein ligase Mdm2For each hit, HMMER outputs a line with several statistics and ending with the name and description of the hit. The first value, the sequence E-value, is the most important as it shows the significance of the hit to the query sequence. The lower this value, the better. The second value is the bit score, a database size-independent metric related to the E-value. The higher, the better. In this section, HMMER shows only one entry per hit, but a hit can have multiple domains matching the query sequence. The statistics in the middle columns refer to the best scoring domain and the number of domains in each hit.
Domain annotation for each sequence (and alignments):
>> 1z1m_A mol:protein length:119 Ubiquitin-protein ligase E3 Mdm2
# score bias c-Evalue i-Evalue hmmfrom hmm to alifrom ali to envfrom env to acc
--- ------ ----- --------- --------- ------- ------- ------- ------- ------- ------- ----
1 ! 218.6 0.1 6.1e-68 1.2e-64 1 110 [] 1 110 [. 1 110 [. 0.99
Alignments for each domain:
== domain 1 score: 218.6 bits; conditional E-value: 6.1e-68
MDM2_MOUSE 1 mcntnmsvstegaastsqipaseqetlvrpkplllkllksvgaqndtytmkeiifyigqyimtkrlydekqqhivycsndllgdvfgvpsfsvkehrkiyamiyrnlvav 110
mcntnmsv t+ga +tsqipaseqetlvrpkplllkllksvgaq dtytmke++fy+gqyimtkrlydekqqhivycsndllgd+fgvpsfsvkehrkiy miyrnlv v
1z1m_A 1 MCNTNMSVPTDGAVTTSQIPASEQETLVRPKPLLLKLLKSVGAQKDTYTMKEVLFYLGQYIMTKRLYDEKQQHIVYCSNDLLGDLFGVPSFSVKEHRKIYTMIYRNLVVV 110
8**********************************************************************************************************976 PPThe details of each hit and all its domains come next. The hit identifier and description are
repeated, preceded by >>, and the domains are sorted by order of appearance in the sequence, not
significance. The c-Evalue and i-Evalue values are conditional and independent E-values. The
first represents the significance of this domain after establishing the hit is a homologue,
while the second measures the significance of the domain if it was the only one being identified.
In general, this latter is the important metric. The next values are the boundaries of the local
alignment, and the last is the expected accuracy per residues of the alignment. The actual domain
alignments make up the remainder of the hit information and include the query sequence (or the
matching fragment), a consensus sequence, and the hit sequence. The last line shows the posterior
probabilities, i.e. the expected accuracy, of each position of the alignment, and can be used to
gauge the less conserved regions of the sequence in the alignment. The last lines of the file show
some statistics on the search itself and on the HMM model building
process.
How many sequences does the RCSB PDB database contain and how many of these matched our query?
Unlike BLAST or the HMMER web server, the local version of HMMER does not provide any sequence
identity or similarity scores. Since these are crucial statistics for deciding on a template for
the modeling, we provide a Python script based on the Biopython library to
parse the sequences and calculate identities. Additionally, the script calculates how much of the
query sequence the hit is matching, also known as coverage. Run the script on the psa.out file
and save the results in a separate psa.info file using the -o option.
python $MOLMOD_BIN/aln_stats.py psa.out -o psa.info
Open and inspect the contents of the alignment information file.
This newly created file aggregates, for each hit, information copied from the HMMER output file and newly calculated values of pairwise sequence identity and coverage. Its simple tabular format is very suitable to identify plausible templates.
#PDBID E-value Bit Score Seq. Id. Seq. Cov. Hit Description
1z1m_A 1.0E-64 218.80 0.90 1.00 Ubiquitin-protein ligase E3 Mdm2
2lzg_A 1.2E-64 218.50 0.90 1.00 E3 ubiquitin-protein ligase Mdm2
2mps_A 2.2E-62 211.30 0.91 0.96 E3 ubiquitin-protein ligase Mdm2
4ode_A 1.8E-60 205.20 0.90 0.95 E3 ubiquitin-protein ligase Mdm2
4odf_A 1.8E-60 205.20 0.90 0.95 E3 ubiquitin-protein ligase Mdm2Apparently, there are plenty of homologues with known structure for mouse MDM2. A quick survey of
the psa.info file shows that more than half of the hits has a sequence identity of at least 90%
to the query sequence, which is the best possible for modeling. Nevertheless, protein structures
are surprisingly robust to changes in sequence. Major structural characteristics are conserved even
at low sequence identities (~30%). As a rule of thumb, any sequence above 30-35% sequence
similarity can be used to build a somewhat
reliable model (source). It is still
possible to model a sequence on a template with 20-30% sequence similarity, the so-called twilight
zone, but there must be extreme care in choosing a proper template. Below 20%, in the danger zone,
there is no guarantee that there is any sequence-structure correlation. Note, though, that all
these are relative percentages to the sequence size.
Choosing a template is, however, more complex than just choosing the most identical homologue. modeling might be a computational method, based on chemical principles, but its fundamental principle is biological. The tertiary structure of a protein defines its function, and as such, folds should be conserved across functionally similar proteins. Consequently, it always pays off to consider the biological function of both the query and the templates and make sure that there is, if possible, a match. Otherwise, nature might have a trick left up in her sleeve (example). In the case of mouse MDM2, most of the hits belong to the E3 ubiquitin ligase family, and several of them are MDM2 proteins of other organisms. It seems then that it is very much possible and straightforward to model mouse MDM2. Finally, the coverage metric also shows that there are very few gaps in the alignment, but it is important to know where these are. If a particular hit has 90% coverage, there could have been an insertion event during evolution. Another possibility, more common, are gaps at one or both termini of the sequences. Either way, gaps are always a point of concern, and there must always be a good justification for building a model from such an alignment.
The second file produced by HMMER (psa.sto) contains the full sequence alignments for each
query/hit pair, which is useful to check where the gaps are in a particular sequence. The Stockholm
format is not as easy to read as FASTA though. Fortunately, HMMER includes a library called easel
whose utilities are very helpful in doing these conversions and manipulations of sequence
(alignment) files.
esl-reformat -o psa.fasta afa psa.sto
Open and inspect the contents of the alignment FASTA file.
The psa.fasta file contains the same information as the Stockholm file, except for the forward
probabilities, just in a different format. The two top scoring hits do not have any gap (coverage
is 1.0). The next few hits have roughly 5% of gaps (coverage 0.95-0.96), which corresponds to ~5
positions in the alignment. Fortunately, these are distributed between the N and C termini of the
protein sequence, or concentrated at the N-terminus. Indeed, even the worst scoring hits have a
consistent region of gaps and a homologous core domain.
>1z1m_A/1-110 [subseq from] mol:protein length:119 Ubiquitin-protein ligase E3 Mdm2
MCNTNMSVPTDGAVTTSQIPASEQETLVRPKPLLLKLLKSVGAQKDTYTMKEVLFYLGQY
IMTKRLYDEKQQHIVYCSNDLLGDLFGVPSFSVKEHRKIYTMIYRNLVVV
>2lzg_A/1-110 [subseq from] mol:protein length:125 E3 ubiquitin-protein ligase Mdm2
MCNTNMSVPTDGAVTTSQIPASEQETLVRPKPLLLKLLKSVGAQKDTYTMKEVLFYLGQY
IMTKRLYDEKQQHIVYCSNDLLGDLFGVPSFSVKEHRKIYTMIYRNLVVV
>2mps_A/1-106 [subseq from] mol:protein length:107 E3 ubiquitin-protein ligase Mdm2
--NTNMSVPTDGAVTTSQIPASEQETLVRPKPLLLKLLKSVGAQKDTYTMKEVLFYLGQY
IMTKRLYDEKQQHIVYCSNDLLGDLFGVPSFSVKEHRKIYTMIYRNLV--
>4ode_A/1-105 [subseq from] mol:protein length:105 E3 ubiquitin-protein ligase Mdm2
-----MSVPTDGAVTTSQIPASEQETLVRPKPLLLKLLKSVGAQKDTYTMKEVLFYLGQY
IMTKRLYDEKQQHIVYCSNDLLGDLFGVPSFSVKEHRKIYTMIYRNLVVV
>4odf_A/1-105 [subseq from] mol:protein length:105 E3 ubiquitin-protein ligase Mdm2
-----MSVPTDGAVTTSQIPASEQETLVRPKPLLLKLLKSVGAQKDTYTMKEVLFYLGQY
IMTKRLYDEKQQHIVYCSNDLLGDLFGVPSFSVKEHRKIYTMIYRNLVVVChoosing a template from the list of homologues
The two closest homologues, as identified by HMMER, have both 90% sequence identity and 100% coverage. The question is thus, which of them should be used to model mouse MDM2? Most homology modeling methods, including MODELLER, can use either one or multiple templates to build the models. Using several different templates is only really advantageous, however, in the case where they provide a better coverage of the query sequence (source). If one template matches the first half of the sequence and another the second half, then both can and should be used to build the full model. Otherwise, a single template with large enough coverage is sufficient to build a high-quality model. Since the N-terminal region of MDM2 that interacts with p53 seems to be a single domain, judging by the consistency of the HMMER results, the best course of action is the use a single template.
As previously mentioned, the main criteria to choose a template are its sequence identity to the query, the coverage, and the biological function. Beyond sequence features and function, it is also important to judge the structural quality of the templates. For example, structures determined by X-ray crystallography differ in the accuracy of the atomic positions in the final structure. On the other hand, NMR structures are built from a collection of experimental (distance, but not only) restraints that can rarely be satisfied by one single conformation. This ambiguity makes NMR structures relatively poor templates, as there is no best model of an ensemble, only one with fewer restraints violations. Additionally, it is not straightforward to discern why a region is floppy in the NMR ensemble: different restraints or a lack of thereof? Unlike NMR structures, X-ray structures have well-defined quality criteria, namely resolution and R-free. The resolution is a measure of the level of detail in the diffraction pattern, which translates to the unambiguity with which atoms fit into the electron density map. Ultra-high resolution structures have values below 1Å. The R-free value reflects the agreement of the final structure with a part of the diffraction data left aside for validation, i.e. a cross-validation measure. An average structure has an R-free value of 0.26, and the lower, the better (source). This preference is of course relative. Between two equally good structures, that determined by crystallography is likely best; however, if the NMR structure is far better in terms of sequence identity, for example, it becomes the obvious template. In this latter case, it might be productive to perform several different single-template modeling runs, each using a different member of the NMR ensemble.
Another factor to consider when choosing a template is its conformation in the crystal structure (or NMR ensemble) and experimental conditions under which the structure was obtained. The presence of co-factors, ligands, and other molecules might have a large impact on the conformation the protein adopts and, therefore, have a direct influence on the choice of using a structure as a template. A possible way of checking the conformational space, or at least its characterized fraction, is to analyse all the released structures related to this particular template. The structural superimposition and subsequent calculation of overall and per-residue root mean square deviations (RMSD) of equivalent atomic coordinates identifies regions that have been crystallised in different conformations and are, therefore, worthy of particular attention when modeling the structure.
To gain some insight on the structural quality of the templates suggested after the HMMER search,
use the RCSB PDB database. The first column of the psa.info file contains
the PDB ID and the PDB chain belonging to the hit sequence. The ID is a four-character
alphanumerical code (e.g. 1z1m) that is unique to each structure, much like the accession code is
for an Uniprot entry. The last character, usually a letter, identifies the chain within the
structure that HMMER identified as a hit.
The three highest scoring templates identified by HMMER (1z1m_A, 2lzg_A, and 2mps_A) are all
NMR structures. The fourth, 4ode_A, is a crystal structure that given its values of sequence
identity (90%) and coverage (95%) is likely the best candidate. The RCSB PDB page for this entry
confirms what HMMER reported: the structure belongs to the E3 ubiquitin-protein ligase MDM2 of
Homo sapiens. Further, the “Experimental Details” and “Structure Validation” sections detail the
high quality of this structure: 1.80Å resolution and above-average R-free and stereochemistry
parameters for structures of similar resolution. The fifth ranked template, 4odf_A, has
virtually the same bit score, identity, and coverage values as 4ode_A. Their very similar names
are also not a coincidence. 4odf_A, as seen in its RCSB PDB entry page, was crystallized by the
same authors, during the same study that produced 4ode_A, using the same experimental methods and
conditions. The only difference is the co-factor that is bound to the protein. The structures
should be, therefore, virtually identical as well.
There are several programs that allow a user to download, visualize, and (quantitatively) compare structures. If possible, use PyMOL, a free and open-source molecular visualization software that runs on Windows, MacOS X, and Linux. For any help with the commands, visit first the community-maintained Wiki and then use Google; chances are that someone else already posted that same problem in the PyMOL mailing-list.
Create a sub-folder (e.g. templates) to store the downloaded structures.
Download and analyse 4ode and 4odf using Pymol.
fetch 4ode, type=pdb1, async=0
fetch 4odf, type=pdb1, async=0
as cartoon
zoom vis
The fetch command of Pymol retrieves a structure from the RCSB PDB database. The async option
enables/disables the asynchronous behavior of the download, or, in other words, if Pymol waits for
the fetch command to finish before executing the next command. We set it to 0, as otherwise the
two last commands will fail. The default set of coordinates downloaded by Pymol is that of the
asymmetric unit (see
here),
the smallest portion of crystal structure to which symmetry operations can be
applied to generate the complete unit cell, which is the crystal repeating unit. Sometimes the
asymmetric unit contains several copies of the structure of interest, which can be tedious to
handle when performing analyses. To circumvent this, the RCSB PDB asks the authors of each crystal
structure to provide their interpretation of a biological unit, or the structure that is actually
functionally active. This can be a simple monomer, or a multi-molecule assembly, such as in the
case of viral capsids. Besides the biological unit assigned by the authors, the RCSB PDB has also
software that automatically detects the biological unit. In the end, specifying type=pdb1 to
Pymol requests the first biological unit of that particular structure, which is usually
appropriate enough. For both 4ode and 4odf, there is only one protein molecule in the
biological unit, which corresponds to chain A. The structures also contain additional
molecules, such as the co-crystallized ligand, solvent, and other small leftovers of the
crystallization process (e.g. SO4). As these will not be needed for the modeling,
remove them. Then, align the structures on each other using the align command.
remove solvent
remove het
align 4odf and name ca+c+n+o, 4ode and name ca+c+n+o, object=aln_4ode_4odf
Coincidentally, the two structures are roughly aligned already from the beginning. The align
command in Pymol performs a sequence-based structure alignment, meaning it superimposes two
structures after aligning their sequences to find equivalent pairs of atoms. As it is written
above, the command will minimize the distances between all equivalent pairs of backbone atoms in
the two structures, using 4ode as a reference. The sequence alignment is saved in a separate
object, aln_4ode_4odf, that can also be visualized to give information on which atoms match which
and where the biggest differences lie. The output of the alignment command is also worthy of
reading:
Match: read scoring matrix.
Match: assigning 99 x 105 pairwise scores.
MatchAlign: aligning residues (99 vs 105)...
ExecutiveAlign: 396 atoms aligned.
ExecutiveRMS: 17 atoms rejected during cycle 1 (RMS=0.26).
ExecutiveRMS: 12 atoms rejected during cycle 2 (RMS=0.22).
ExecutiveRMS: 6 atoms rejected during cycle 3 (RMS=0.20).
ExecutiveRMS: 3 atoms rejected during cycle 4 (RMS=0.20).
ExecutiveRMS: 2 atoms rejected during cycle 5 (RMS=0.19).
Executive: RMS = 0.192 (356 to 356 atoms)
Executive: object "aln_4ode_4odf" created.Why is there a different number of residues between the two structures?
The align command aligns the minimum number of common atoms (99 x 4 = 396) between the structures
and iteratively removes outliers, those that contribute the most to the RMS value, in order to
optimize the superimposition. The default number of cycles is 5, but this can be controlled with
the cycles option (e.g. cycles=1). This is sometimes useful to obtain a proper full-structure
RMS value, since there is no control over which atoms are effectively removed; i.e. different pairs
of structures can, and will likely have, different outliers that make comparisons between RMS
values wrong. In this case, since there is only one comparison to make and the initial RMS is
already extremely low (0.26Å), controlling the number of cycles is a futile exercise.
Besides observing and analyzing the structures, Pymol provides information about the
superimposition in the sequence viewer. To access it, click the S icon on the bottom right
corner of the viewer window or click on Display -> Sequence on the menu bar. The residues are
represented in one-letter notation and gaps in the sequence alignment (if the object is active) are
represented as dashes. Residues whose atoms have been removed during the iterative optimization of
the superimposition are colored gray. This allows users to quickly check which regions are not
matching. Note though that the align command performs a pairwise sequence alignment, and that if
you have done multiple alignments you should check each one separately.
Either way, the quick analysis shows that 4ode and 4odf are similar not only in name but also
in conformation. In fact, they are virtually identical with an RMS below 0.5Å!
Repeat the same analysis for the top hit of HMMER: 1z1m
Having concluded that 4ode is a suitable template to model mouse MDM2, save the structure
(without solvent or heteroatoms) from Pymol to a new PDB file.
Another common issue with PDB files is the presence of atoms with multiple occupancies, or atoms whose position could not be unambiguously determined in the electron density map. Modeling programs usually complain, sometimes loudly, about these and they add little to no information to the modeling protocol itself (although they can be useful in some specific scenarios!). Usually, it suffices to keep the highest occupancy position of each atom.
Using the pdb-tools suite, clean the 4ODE_A PDB file of multiple occupancies.
pdb_delocc.py 4ODE_A.pdb > 4ODE_A.pdb.clean
mv 4ODE_A.pdb.clean 4ODE_A.pdb
As a side note, choosing one single template might not be ideal in all cases. When modeling a multi-domain protein, for example, or when a single domain is not complete in any template, it is wise to use multiple templates simultaneously to build the model. Some also defend that using multiple templates for a single domain might help eliminating errors from the crystal structures, although this is disputed. Regardless, this is beyond the scope of this course. Have a look here if you want to explore this issue further.
Modeling mouse MDM2 using MODELLER
Having chosen a template, it is time to focus on building the model itself. While there are many homology modeling methods in the literature, MODELLER, developed by Andrej Šali and a host of co-workers since the early 1990s, stands out as a robust and popular software package. It builds a structural model from a set of automatically generated spatial restraints, but it also allows the user to define other types of restraints such as secondary structure definitions, generic distance restraints from NMR or cross-linking mass spectrometry, angle and dihedral angle restraints, and also Cryo-EM density maps. If you wish to learn more about MODELLER, have a look at the online manual and the tutorial pages.
To run, MODELLER requires an alignment file with the sequence to be modelled, in our case mouse MDM2, and the sequence(s) of the template(s). In addition, it obviously requires the structure(s) of the template(s). The input alignment file MODELLER reads must be in a modified PIR format. This format is quite unusual and requires particular attention to write correctly. From experience, the overwhelming majority of the errors MODELLER throws at the user come from inconsistencies in the alignment. For an in-depth description of this format, read the MODELLER manual webpage.
# Example PIR alignment
>P1;5fd1
structureX:5fd1:1 :A:106 :A:ferredoxin:Azotobacter vinelandii: 1.90: 0.19
AFVVTDNCIKCKYTDCVEVCPVDCFYEGPNFLVIHPDECIDCALCEPECPAQAIFSEDEVPEDMQEFIQLNAELA
EVWPNITEKKDPLPDAEDWDGVKGKLQHLER*
>P1;1fdx
sequence:1fdx:1 : :54 : :ferredoxin:Peptococcus aerogenes: 2.00:-1.00
AYVINDSC--IACGACKPECPVNIIQGS--IYAIDADSCIDCGSCASVCPVGAPNPED-----------------
-------------------------------*HMMER produces a local sequence alignment between the query and each hit, which is not useful
for the modeling since it might not cover the entire query sequence. However, it does produce a
file containing the full aligned sequence of the hits, including the 4ode template sequence,
which you have previously converted to a FASTA format: psa.fasta.
Your alignment.pir file should now be looking like this:
MCNTNMSVSTEGAASTSQIPASEQETLVRPKPLLLKLLKSVGAQNDTYTMKEIIFYIGQYIMTKRLYDEKQQHIVYCSNDLLGDVFGVPSFSVKEHRKIYAMIYRNLVAV
-----MSVPTDGAVTTSQIPASEQETLVRPKPLLLKLLKSVGAQKDTYTMKEVLFYLGQYIMTKRLYDEKQQHIVYCSNDLLGDLFGVPSFSVKEHRKIYTMIYRNLVVVThe first line, the mouse MDM2 sequence, needs to be reformatted into the format shown above in the
example PIR alignment, as well as moved to the bottom of the file. Before each sequence, there must
be a header line with a meaningful name, such as MDM2_MOUSE for the query or 4ODE_A for the
template. The identifier for the template must match the PDB file name (except the extension).
The identifier for the query sequence will be used also when creating the structural models. The
second line starts with sequence and is followed by several fields separated by colons (:). The
fields are explained in detail in the description of the format, so do take some time to read it.
The first field is the name of the sequence, matching the line before; the second and fourth are
the first and last residue numbers of the sequence, usually 1 and the length of the sequence. The
remaining fields are optional. The remaining lines are for the sequence itself. It can span as many
lines as necessary or aesthetically pleasant, although it is suggested is to keep the lines around
80 characters long. The sequence must be terminated with an asterisk symbol (*). The template
sequence follows a similar pattern, except it indicates structureX at the beginning of the second
line and the residue numbering fields have to match the PDB file. Additionally, as RCSB deposited
structures are properly formatted (mostly anyway), the third and fifth fields must contain the
chain identifiers that are to be used in the modeling. Again, the name of the structure must
match the name of the PDB file without the extension, so pay special attention to that.
>P1;4ODE_A
structureX:4ODE_A: 6: A: 110: A::::
-----MSVPTDGAVTTSQIPASEQETLVRPKPLLLKLLKSVGAQKDTYTMKEVLFYLGQYIMTKRLYDEKQQHIVYCSNDLLGDLFGVPSFSVKEHRKIYTMIYRNLVVV*
>P1;MDM2_MOUSE
sequence:MDM2_MOUSE: 1: : 110: ::::
MCNTNMSVSTEGAASTSQIPASEQETLVRPKPLLLKLLKSVGAQNDTYTMKEIIFYIGQYIMTKRLYDEKQQHIVYCSNDLLGDVFGVPSFSVKEHRKIYAMIYRNLVAV*Using MODELLER requires some programming knowledge. The software exposes a very complete Python API that allows users to create (simple) scripts to control all parameters of the modeling protocol. There are plenty of example scripts on the documentation and tutorial pages of MODELLER, but for your convenience, we provide one.
Start the modeling process using the cmd_modeller.py script. Keep on reading while it runs.
cmd_modeller.py -a alignment.pir -t 4ODE_A.pdb --use_dope --num_models 10
The protocol and settings the scripts will use are what our group uses in real modeling jobs in
the lab. By default, it uses the MODELLER routine automodel to generate models, which automates
most of the model building protocol, including a small refinement step. MODELLER starts by reading
and validating the alignment against the PDB file(s) of the template(s). If a PDB file is missing
some fragment of the sequence HMMER retrieved for that template, for example because it could not
be observed in the electron density, then the alignment will have to be manually corrected.
MODELLER is quite verbose when it comes to these and other error messages. In this particular case,
it will show exactly where the discrepancy occurs. To avoid multiple iterations of trial and error,
simply extract the sequence directly from the ATOM lines of the PDB file using the utility script
pdb_toseq.py and align it to the sequence given by HMMER using for example the global pairwise
alignment algorithms hosted at the EBI servers. This will
highlight any missing regions.
Why would some regions be missing in the electron density map of a crystal structure?
The next step in the modeling protocol is to calculate the coordinates of the atoms of an initial
model. Equivalent atoms between query and template will be simply copied, and in the case of
multiple templates, their positions averaged over all templates. The remaining atoms will be built
from scratch using internal coordinates and the CHARMM topology library. Afterwards, MODELLER will
create all the spatial restraints it will use to refine the model. These include, but are not
limited to, stereochemical (bonds, angles, dihedrals, impropers) and homology-derived (distances
between residues) restraints. The stereochemical restraints are derived from statistical analyses
of many pairs of homologous structures. For each requested model, MODELLER will apply an
optimization algorithm to fit the model as best as possible to all the restraints. Each model will
be slightly randomized before this optimization, so that there is some variability at the end of
the protocol. The optimization is done in several steps, first taking into account only restraints
between atoms close in sequence, and later all other restraints. The optimization is carried out
via a combination of conjugate gradients and molecular dynamics with simulated annealing. All
models are then evaluated according to their stereochemical quality and the degree of restraints
violations - the molpdf score. The resulting PDB files end in .B9999*.pdb. MODELLER also
includes other scoring functions to gauge the quality of the models and their resemblance to
native structures, such as the DOPE energy potential, which was derived from existing structures.
Once MODELLER is finished, it will produce a listing of the models it created together with the values of whichever scoring functions we asked it to include. The models are not ranked by energy or quality, but by filename. The following is an excerpt of the models produced from the 4ODE template for the 1-110 region of the MDM2 mouse sequence.
>> Summary of successfully produced models:
Filename molpdf DOPE score
-------------------------------------------------------
MDM2_MOUSE.B99990001.pdb 542.16193 -12232.60254
MDM2_MOUSE.B99990002.pdb 493.51770 -12263.11816
MDM2_MOUSE.B99990003.pdb 618.48962 -12130.24023
MDM2_MOUSE.B99990004.pdb 538.61859 -12210.15723
MDM2_MOUSE.B99990005.pdb 625.59253 -12057.88477
MDM2_MOUSE.B99990006.pdb 687.50299 -12036.90625
MDM2_MOUSE.B99990007.pdb 565.85083 -12211.46484
MDM2_MOUSE.B99990008.pdb 587.98169 -12133.75293
MDM2_MOUSE.B99990009.pdb 566.93512 -12212.98145
MDM2_MOUSE.B99990010.pdb 549.83215 -12059.09277Do the molpdf and DOPE scores correlate?
Compare the three best models (by DOPE score) and the template structure in Pymol.
pymol 4ODE_A.pdb MDM2_MOUSE.B99990002.pdb MDM2_MOUSE.B99990001.pdb MDM2_MOUSE.B99990009.pdb
Overall, the models are virtually identical and also identical to the template. This is not
surprising, given the high degree of identity between the query and template sequences and the fact
that the template sequence covers nearly all of the query. The DOPE and molpdf scores are also
not very informative. The different models differ very slightly, particularly in DOPE score.
Interestingly, the two scores are not correlated, which is again not surprising since they evaluate
different properties. The molpdf score only informs about the agreement of the model with the
restraints derived from the alignment, while the DOPE score tries to inform on the likelihood of
the model resembling a real structure. An important feature of MODELLER and its scoring scheme is
the possibility of obtaining per-residue scoring profiles, namely of the DOPE potential, which
allow the identification of regions of the model that need further care. By convention, any residue
scoring above -0.030 is considered problematic. However, keep in mind that the DOPE potential is
not as fine-grained to single out badly modeled residues. Additional modeling should only be
considered if a stretch of several residues scores consistently near or above this threshold.
Build per-residue profiles of different models and inspect them visually.
evaluate_model.py MDM2_MOUSE.B99990002.pdb
plot_profile.py MDM2_MOUSE.B99990002.dope_profile
There are many possible strategies to improve the (local) quality of a protein model. In this
particular case, the model is particularly worse at the N-terminal region, which correlates with
the lack of structural information in the template. MODELLER has specific protocols that address
such regions, called loops. By selecting the loopmodel routine instead of the default
automodel, after building the backbone models, MODELLER will proceed to build the loops if there
are any gapped regions in the alignment. The atoms in these regions are placed in a line connecting
the carbonyl oxygen and amide nitrogen of the flanking (known) residues and then their conformation
is refined using an atomistic distance-dependent statistical potential for non-bonded interactions.
A second refinement step takes place, in context of the rest of the protein, that is, with the loop
atoms feeling the rest of the protein. The final models end in .BL*.pdb and are also scored
using the same molpdf score, by default, and any other additional scoring functions. Keep in
mind, however, that the accuracy of the loop modeling protocol degrades (very) rapidly with the
loop length.
Redo the modeling using the loop modeling routine.
The loop models are also ranked with both the molpdf and DOPE scores. Given the complete freedom
of the loop atoms to sample the conformational space – unlike the rest of the protein, there are
no alignment-based restraints – it is no surprise that first, the molpdf scores are much lower,
and second, there is much more energetic discrepancy between the different models. Also, at first
glance, the DOPE score of the models worsened drastically. However, the scores reported in the loop
model listing reflect only the loop region. To obtain a full model DOPE score, use the
evaluate_model.py script and note the score given at the end.
>> Summary of successfully produced models:
Filename molpdf DOPE score
-------------------------------------------------------
MDM2_MOUSE.B99990001.pdb 542.16193 -12232.60254
>> Summary of successfully produced loop models:
Filename molpdf DOPE score
-------------------------------------------------------
MDM2_MOUSE.BL00010001.pdb -4.57528 -442.76462
MDM2_MOUSE.BL00020001.pdb -29.39576 -553.64691
MDM2_MOUSE.BL00030001.pdb 8.09913 -308.24115
MDM2_MOUSE.BL00040001.pdb -7.04052 -560.09100
MDM2_MOUSE.BL00050001.pdb 8.53783 -410.75394
MDM2_MOUSE.BL00060001.pdb -0.54786 -578.25775
MDM2_MOUSE.BL00070001.pdb -25.18821 -989.38574
MDM2_MOUSE.BL00080001.pdb -22.06119 -512.21118
MDM2_MOUSE.BL00090001.pdb -21.29531 -534.20166
MDM2_MOUSE.BL00100001.pdb 15.27596 -294.98157Compare the three best loop models (by DOPE score) and the backbone model in Pymol.
Did the loop modeling protocol significantly improve the scores of the N-terminal loop? Why so?
This validation using the tools bundled with MODELLER helps understand the limitation of the models. Analyzing which regions are reliable and which are more likely to be incorrect is an extremely important part of the modeling exercise, particularly before handing out the model to collaborators or using it to draw any functional/biological conclusions. Besides the DOPE profiles, there are other dedicated validation protocols that analyze the quality of the models based on many different criteria. Many of these are available as web servers, such as QMEAN, PSVS and Molprobity. These servers report on both the overall quality of the model and per-residue profiles, using metrics based on statistical comparisons to existing high-resolution crystal structures. For instance, they calculate all bond lengths and angles in the model and compare the distribution with that found in experimental structures. Using these dedicated validation servers is a quick and reliable way of checking the quality of one or more homology models, and is usually advised in any realistic Modeling application.
pymol 4ODE_A.pdb MDM2_MOUSE.BL00070001.pdb
align MDM2_MOUSE.BL00070001, 4ODE_A
zoom vis
show cartoon
Congratulations!
You started with a sequence of a protein and went all the way from finding possible templates, to evaluating which to use, to building several models, assessing their quality, and finally selecting one representative. This model can now be used to offer insights on the binding of MDM2 to p53, or on the structure of the mouse MDM2 protein, or to seed new computational analysis such as docking.
You might want to continue with the tutorial on molecular dynamics simulations!