

Using AlphaFold to model the MDM2-p53 complex
General Overview
This extra module provides an introduction on how artificial intelligence algorithms are being used in the field of computational structural biology and guides you on how the use of AlphaFold to predict macromolecular complexes.
Introduction
DeepMind Technologies is an artificial intelligence subsidiary of Alphabet Inc. (Google). It broke the news in 2016 by presenting AlphaGo, a computer program that plays the board game Go, and managing to beat the world’s top player Lee Sedol (news article. This competition has also been made into a movie). Some might say that Go is the most complex game in the world due to its vast number of variations in individual games and because its large board and lack of restrictions allow for a great variation in strategies expressing the players’ individualities. Actions made in one part of the board may be influenced by an apparently unrelated situation in a distant part of the board.
Later on, in 2019 DeepMind unveiled AlphaStar, a computer program designed to play the strategy computer game Starcraft II. This was also considered a major breakthrough since StarCraft is a real-time game (not turn-based) with partial information (you cannot see the entire board, the map, at once) with no single dominant strategy and complex rules. This was met with some controversy by top-players with allegations that AlphaStar had an unfair advantage because it could click faster than a human. :)
In their most recent effort, AlphaFold, DeepMind tackled the challenging protein structure prediction problem. The complexity of this problem is illustrated by the Levinthal Paradox, firstly proposed in 1969 by the molecular biologist Cyrus Levinthal:
(…) each bond connecting amino acids can have several (e.g., three) possible states, so that a protein of, say, 101 amino acids could exist in \(3^{100} = 5\times10^{47}\) configurations. Even if the protein is able to sample new configurations at the rate of \(10^{13}\) per second, or \(3 \times 10^{20}\) per year, it will take \(10^{27}\) years to try them all. Levinthal concluded that random searches are not an effective way of finding the correct state of a folded protein. Nevertheless, proteins do fold, and in a time scale of seconds or less. This is the paradox.
Nature is extremely efficient at optimizing processes. Nature’s solution to the Levinthal’s paradox allowed complex life to exist as not all of the aforementioned \(5x10^{47}\) possible conformations would be thermodynamically stable and biologically functional. Protein folding is mainly guided by an hydrophobic collapse and the formation of a hydrogen bond network, then stabilized by van der Waals forces and electrostatic interactions and hence follow a folding path which is not random. This is also known as the Thermodynamic Hypothesis. There are several experimental techniques to study protein folding such as fluorescence spectroscopy, circular dichroism and of course Nuclear Magnetic Resonance (NMR) spectroscopy. On the computational side of things, ab initio (or de novo) protein structure prediction software may aim to obtain the folded state of a protein by simulating the folding proceess via long molecular dynamics simulations, in a process analogous to what is observed in nature. This is however a computationally expensive process, limited therefore to rather small proteins and usually tackled by large-scale computational projects such as Rosetta@home and Folding@home or using custom-build computers like Anton, a line of supercomputers, designed and built by D.E. Shaw Research (DESRES) specifically for running molecular dynamics simulations. Alternatively different heuristics can be applied that commonly use information from experimentally solved structures such as for example: Fragment-based methods (Rosetta), evolutionary co-variation (EVFold) and protein threading (I-TASSER).
Machine Learning
According to the Thermodynamic Hypothesis all the information that governs how proteins fold is contained in their respective primary sequences. This paradigm was the catalyst for the development of several computational algorithms to score protein conformations in the search of the lowest free energy state (also named the native state). The main issue with this energy-driven approach is that the search space size is a function of the protein’s length. The common approach for protein structure prediction is to use intermediate simplified steps that reduce the complexity of the structure but retain information. This is called Protein Structure Annotation (PSA). PSAs can be 2-dimensional (secondary structure elements, contact maps) or 1-dimensional (e.g. solvent accessibility, and torsional angles). With the steady growth of the number of experimentally determined protein structures in the PDB, an increasing number of PSAs are available. These annotations can then be used in a variety of deep learning methods, such as AlphaFold and RoseTTAFold.
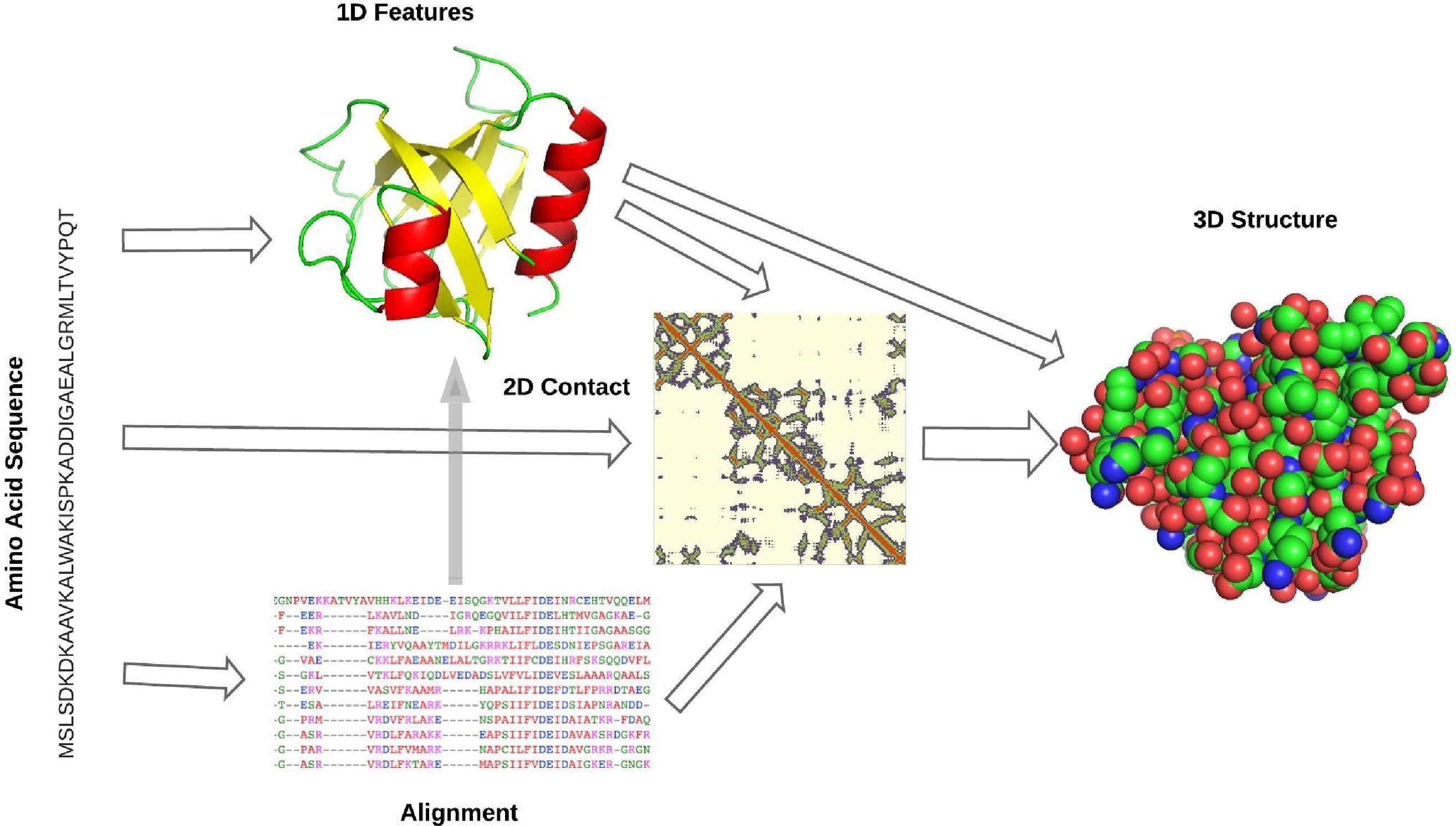
Deep learning is a sub-field of machine learning which is based on artificial neural networks (which may seem like a new thing but it was first thought of in the 40’s!). It uses multiple connected layers to transform inputs (PSAs in our case) into features that are then used to predict a corresponding output. Neural networks attempt to simulate the behavior of the human brain allowing it to “learn” from large amounts of data. A neural network with a single layer can still make approximate predictions, but additional hidden layers (deep learning) can help to optimize and refine for accuracy.
If you are interested in the field of machine learning, refer to the links at the bottom of the page for a starting point in your AI journey since an in-depth explanation of machine learning is out of the scope of this course. In this extra module we will focus solely on the use and application of AlphaFold. Keep in mind that despite being famous for both its astonishing performance (discussed below) and great marketing department, AlphaFold is not the only such software; other researchers have been applying neural networks to the issue of protein folding such as shown in this article in Cell that uses a recurrent geometrical network.
AlphaFold 2
Briefly put, DeepMind trained AlphaFold on more than 170 000 protein 3D structures available in the the protein data bank. It compared multiple sequences in the data bank and looked for co-evolving pairs of residues that often end up close together in the folded structure. It then used these data to guess the distance between pairs of amino acids in structures that are not yet known. The training of the model took “a few weeks”, using computing power equivalent to between 100 and 200 GPUs - graphical processing units (or video cards). The model uses a form of attention network, which is a deep-learning technique that allows the program to focus on smaller parts of a larger problem. The code if freely available on GitHub, but be aware that installing the full model requires over 1TB of disk space!
Check DeepMind’s article published on Nature in 2021: “Highly accurate protein structure prediction with AlphaFold”
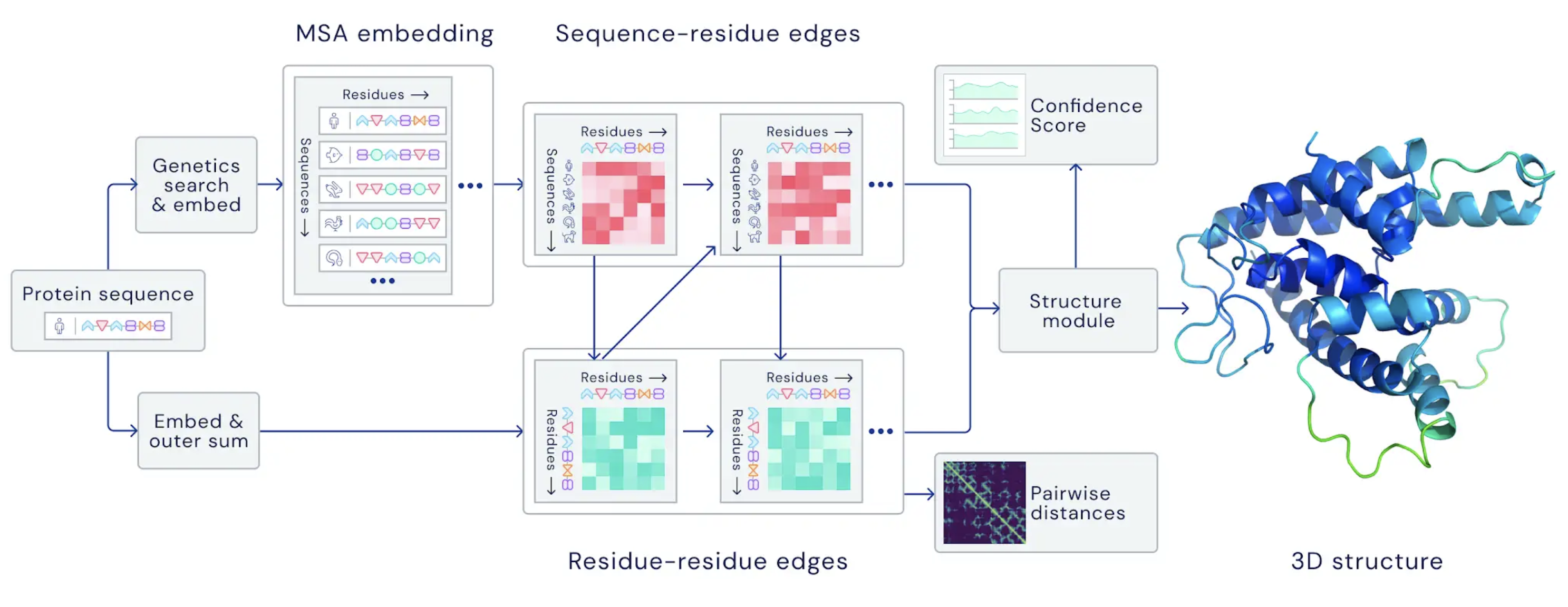
This image is an overview of the main neural network model architecture. The model operates over homologous protein sequences as well as amino acid residue pairs, iteratively passing information between both representations to generate a 3D structure (source: DeepMind).
Given this introduction, what is the buzz all about? Many of us saw the news that AlphaFold solved the folding problem, which can be easily misinterpreted. The folding problem refers to the process, i.e. how the amino-acid chain generated by the ribosome reaches its final stable state (the “folder” or “native” state). It thus refers to the thermodynamic pathway that a protein takes to get to its native state. There have been discussions in the community that current protein structure predictors do not produce meaningful folding pathways, but this not what they are originally trained for. These methods, however, including AlphaFold, provide the necessary tools to obtain the 3D structure of many proteins that would not be feasible via ab initio or comparative modelling techniques.
This was nicely demonstrated by the results obtained by AlphaFold in the Critical Assessment of protein Structure Prediction (CASP). This assessment is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. It functions as a benchmark in the field of protein prediction. In every round, CASP provides research groups with one or more targets, which are the sequences of proteins that has been solved experimentally but are not yet available publicly. The research groups must then apply their methods to predict the structure of this protein and by the end of the round, their predictions are compared to the experimental structure.
For two years in a row (2018 and 2020), DeepMind’s AlphaFold was ranked first in CASP, with a prediction accuracy well beyond that of any other group. In the latter edition, AlphaFold generated the best prediction for 88 out of the 97 targets, achieving a median accuracy score of 92.4 (out of 100) in the global distance test, a level of accuracy that is equivalent to experimental techniques…! During the 2020 CASP conference experimentalist groups working with particularly challenging complexes were contacted and it was suggested that they should use the protein structures predicted by AlphaFold to aid in the experimental structure determination (with a technique called molecular replacement). Surprisingly, by using AlphaFold predictions researchers were able to experimentally determine structures that were so far resilient to structure determination. You can read their very enthusiastic responses in the presentation slides).
Predicting the MDM2-p53 complex with AlphaFold
In the previous modules of this tutorial you have been guided trough the comparative modelling of MDM2, the generation and sampling of p53 peptide conformations and, finally, molecular docking of those structures.
While AlphaFold was initially built for the prediction of monomers, scientists do enjoy pushing things to the limit and started testing several scenarios for which AlphaFold was not trained. This resulted in a series of “spin-offs”, the most relevant one in the context of this tutorial being AlphaFold-Multimer (not yet peer reviewed).
AlphaFold-Multimer’s goal is to predict the 3D structure of molecular complexes. It was tested on 4443 complexes and successful predictions were obtained for 67% of the cases with heteromeric interfaces and for 69% of cases with homomeric interfaces.
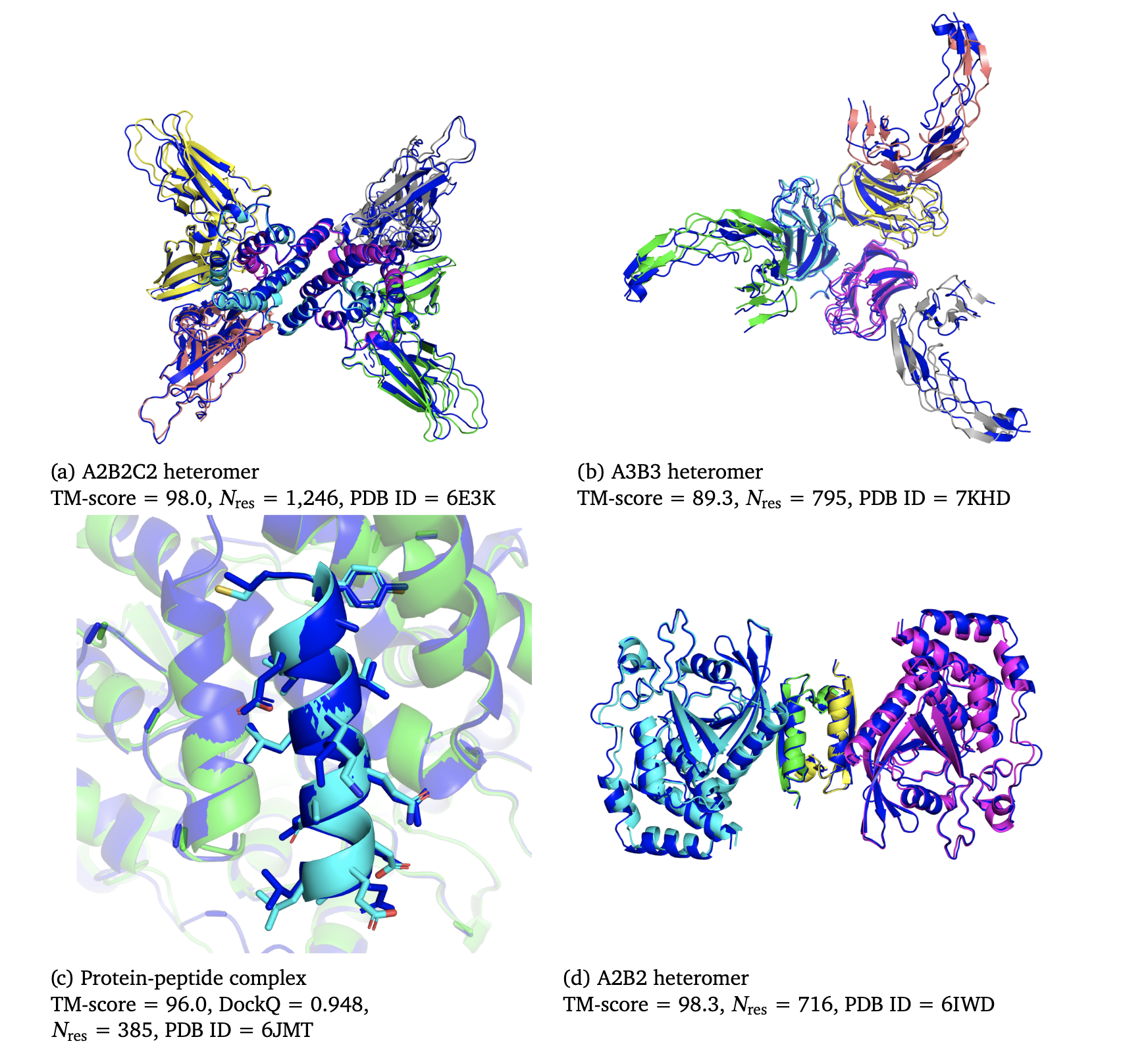
This figure from their paper shows examples predicted with the AlphaFold-Multimer. In blue are the native structures and the predictions are colored by chain (source: DeepMind).
In the picture above (c) you can see a predicted Protein-Peptide complex that is extremely similar to the experimental structure. The natural question is: Could we use AlphaFold to predict the MDM2-p53 complex?
Accessibility and ease of use are important factors for every computer software and the installation and setup of AlphaFold is not trivial. This requires the download of a large database (2TB) and a lot of processing power, unfortunately not something that can be executed on a laptop. To address this, AlphaFold has been made available for “free” (since you need a Google account) within the Colab platform which “Allows you to write and execute Python in your browser, with [z]ero configuration required, [f]ree access to GPUs.”.
The official AlphaFold Colab has a limit on the minimum number of residues you can input, so we will use instead a community-made version that is slightly tweaked but is sufficient for our protein-peptide study.
Click here to go to ColabFold and read the description.
Input the sequences of our complex in the query_sequence.
Note that you will need to add a : between the two sequences of the components of the complex:
MCNTNMSVSTEGAASTSQIPASEQETLVRPKPLLLKLLKSVGAQNDTYTMKEIIFYIGQYIMTKRLYDEKQQHIVYCSNDLLGDVFGVPSFSVKEHRKIYAMIYRNLVAV:SQETFSGLWKLLPPE
In the top section of the Colab, click: Runtime > Run All
(It may give a warning that this is not authored by Google, because it is pulling code from GitHub). This will automatically install, configure and run AlphaFold for you - leave this window open. After the prediction complete you will be asked to download a zip-archive with the results.
Now that you are quite literally on the edge of the computational structural biology field, try to answer these (difficult) questions:
How do you interpret AlphaFold’s prediction? What is the predicted LDDT?
How can you visualize the accuracy of the predicted model?
Tip: Try to find information about the prediction confidence at https://alphafold.ebi.ac.uk/faq and use that information to visualize the prediction confidence of the model in PyMol
See tips on how to visualize the prediction confidence in PyMol
To color the complex by-chain and identify the position of the peptide: util.cbc When looking at the structures generated by AlphaFold in PyMol, the pLDDT is encoded as the B-factor. Analyze what is the pLDDT of prediction around the interaction interface. To color the model according to the pLDDT type in PyMol: spectrum b
How do the models (docking and AlphaFold) compare to the human complex? Which one is “best”?
It might be frustrating realizing how easy and how fast it is to predict the MDM2-p53 complex with AlphaFold. This case is a very well-studied complex so there is plenty of information about it to drive AlphaFold’s algorithm, however this is not the case for all structures.
While this is without the shadow of a doubt a major breakthrough, it is not an answer to all the modelling problems of the world (… but maybe to most..!). The knowledge of how to do comparative modelling, molecular dynamics simulation, and docking allows you do critically judge the predictions, understand its advantages, and identify its limitations.
Congratulations!
You reached the end of this extra module and got a brief overview on how artificial intelligence methods are being used in the field of computational structural biology and used AlphaFold to model the MDM2-p53 complex.
If you want to read more about the potential of AlphaFold for predicting the structure of protein-peptide complexes, refer to this Nature Comm. paper.