

HADDOCK2.2 manual
Analysis
The analysis of the docking results are performed after the semi-flexible simulated annealing step and after the explicit solvent refinement. A number of standard CNS analysis scripts are automatically run by HADDOCK and the results are placed in the analysis directory in runX/structures/it1 and runX/structures/it1/water, respectively. Some of the generated output files are parsed automatically by HADDOCK to generate for example violations statistics (see violation analysis). Another important step consists in a manual analysis of the generated structures and their clusters. This is the critical step for the classification of the docking solutions and the identification of the best(s) cluster(s).
Topics:
- Standard analysis performed by HADDOCK
- Average structure and RMSDs
- pairwise RMSD matrix
- Energy and buried surface area analysis
- Desolvation energy analysis
- Per-residue interaction energy
- Covalent geometry analysis
- Distance restraints (AIRs, unambig, Hbonds) violations analysis
- Dihedral angle restraints violations analysis
- Residual dipolar coupling restraints violations analysis
- Intervector projection angle restraints violations analysis
- Diffusion anisotropy restraints violations analysis
- Intermolecular hydrogen bonds analysis
- Intermolecular hydrophobic contacts analysis
- Violation analysis
- Manual analysis
- Rerunning the analysis for a given cluster
Standard analysis performed by HADDOCK
The following CNS analysis scripts are automatically run by HADDOCK:
- Average structure and RMSDs
- Pairwise RMSD matrix
- Energy and buried surface area analysis
- Desolvation energy analysis
- Per-residue interaction energy
- Covalent geometry analysis
- Distance restraints (AIRs, unambig, Hbonds) violations analysis
- Dihedral angle restraints violations analysis
- Residual dipolar coupling restraints violations analysis
- Intervector projection angle restraints violations analysis
- Diffusion anisotropy restraints violations analysis
- Intermolecular hydrogen bonds analysis
- Intermolecular hydrophobic contacts analysis
- Note1: If less than three atoms are selected when using the defined semi-flexible
segments, then the entire backbone will used. If still less than three atoms are selected,
then all heavy atoms will be used for the fitting. This makes sure that at least three atoms
are selected for any molecule, including small ligands.
Output files:
- fileroot_ave.pdb: average structure
- filerootfit_1.pdb, filerootfit_2.pdb, ...: superimposed structures
Note2: The numbering of the superimposed PDB files does not correspond with the numbering in the it1 or water directories, but to the position of the structure in the sorted file.list file, i.e. structure number 1 in the analysis directory is the first (best) in file.list and structure number 50 is at position 50 in that file.
- rmsave.disp: contains the RMSD from the average structure for each structure and
the average values over the ensemble. For this, the structures are superimposed on the backbone
atoms of the flexible interface (see Note1 above) and the following average RMSD
values from the average structure are calculated and written to file:
- RMSD backbone interface of all molecules
- RMSD complete backbone of all molecules
- backbone interface of molecule A
- backbone interface of molecule B
- backbone interface of molecule C
- ...
In addition to the average RMSD calculated from the entire ensemble, the corresponding single structure RMSD values are listed in rmsave.disp
- rmsdseq.disp: per residue RMSDs (backbone heavy atoms (N,CA,C), extended backbone heavy atoms
(N,CA,CB,C,O), side-chain heavy atoms and all heavy atoms.
- fileroot-reduced.crd: trajectory file containing only the coordinates of the flexible interface backbone atoms (see Note1 above); this reduced file is used to calculate the pairwise RMSD matrix and thereby speed up the calculations.
Output files:
- fileroot_rmsd.disp: this file contains the pairwise RMSD matrix with on each line three number:
the structure numbers of the two structures being compared and the corresponding RMSD value.
Note5: The numbering of the structures corresponds to the position of the structure in the sorted file.list file.
This file is used as input for the RMSD clustering.
- over the entire complex
- over the flexible interface only (as defined in the run.cns parameter file)
- only the intermolecular energies (vdw and elec)
Output files:
- energies.disp: this file contains the various energy terms per structure and averaged over the ensemble
- Complex statistics: Etot, Ebond, Eangle, Eimpr, Edihed, Evdw, Eelec
- Flexible interface statistics: Etot, Evdw, Eelec
- Intermolecular statistics: Etot, Evdw, Eelec
- Buried surface area
Output files:
- edesolv.disp: this file contains the desolvation energy per structure and averaged over the ensemble
Output files:
- ene-residue.disp: this file contains the various energy terms per structure and averaged over
the ensemble
Example:
#Residue ASP 38 A - intermolecular energies
#file Etot Evdw Eelec
# PREVIT:e2a-hpr_161.pdb -16.7601 -3.41526 -13.3448
# PREVIT:e2a-hpr_189.pdb -42.4061 -1.83788 -40.5682
...
# mean values for interaction with residue ASP 38 A
# ASP 38 A : Etot -34.528 (+/- 21.4012 ) [kcal/Mol]
# ASP 38 A : Evdw -1.34906 (+/- 0.967306 ) [kcal/Mol]
# ASP 38 A : Eelec -33.179 (+/- 21.1375 ) [kcal/Mol]
...
The average per-residue values can be easily extracted from this file and sorted in decreasing contribution with the following command:
grep ": Evdw" ene-residue.disp |sort -gk7 grep ": Eele" ene-residue.disp |sort -gk7 grep ": Etot" ene-residue.disp |sort -gk7
Output files:
- geom.disp: this file contains the averaged deviations from ideal geometry per structure and
averaged over the ensemble.
- print_geom.out: this file contains the listing of covalent terms deviating from the ideal geometry:
- bonds > 0.025 A
- angles > 2.5 degrees
- improper dihedrals > 2.5 degrees
- dihedral angles > 30 degrees
Output files:
- noe.disp: this file contains the number of distance restraints violations per
structure and averaged over the ensemble over all distance restraint classes and for each
class (unambiguous, ambiguous, hbonds) separately. Distance restraints violation > 0.5, 0.3
and 0.1 A are reported.
- print_dist_all.out: this file contains the violation listing for all distance restraints including hbond restraints.
- print_dist_noe.out: this file contains the violation listing for all distance distance restraints
(unambiguous and ambiguous classes).
- print_noe_unambig.out: this file contains the violation listing for the unambiguous
distance restraints.
- print_noe_ambig.out: this file contains the violation listing for the ambiguous distance
restraints (typically the class used to define Ambiguous Interaction Restraints).
- print_dist_hbonds.out: this file contains the violation listing for the hydrogen bond distance restraints.
- Note6:: The above five files (print_....out) are parsed automatically by HADDOCK to
generate statistics on a restraint basis over all structures in the ensemble
using the ana_noe_viol.csh script provided in the tools directory (see
violation analysis).
Output files:
- dihedrals.disp: this file contains the number of dihedral restraints violations per structure
and averaged over the ensemble.
- print_dih.out: this file contains the violation listing for all dihedral restraints. This file is parsed automatically by HADDOCK to generate statistics on a restraint basis over all structures in the ensemble using the ana_dihed_viol.csh script provided in the tools directory (see violation analysis).
Output files:
- sani.disp: this file contains the number of dipolar coupling violations per structure and
averaged over the ensemble.
- print_sani.out: this file contains the dipolar couplings violation listing. (No automatic parsing of this file is currently implemented).
Output files:
- vean.disp: this file contains the number of intervector projection angle restraints violations
per structure and averaged over the ensemble.
- print_vean.out: this file contains the intervector projection angle restraints violation listing. (No automatic parsing of this file is currently implemented).
Output files:
- dani.disp: this file contains the number of diffision anisotropy violations per structure and averaged over the
ensemble.
- print_dani.out: this file contains the diffision anisotropy violation listing. (No automatic parsing of this file is currently implemented).
Output files:
- hbonds.disp: this file contains a listing of all intermolecular hydrogen bonds over the ensemble of
structures. It is automatically parsed by HADDOCK using the ana_hbonds.csh script located
in the tools directory. This scripts generate a listing (ana_hbonds.lis) of
intermolecular hydrogen bonds including the number of occurrences and the average hydrogen bond
distance.
-
Note7: The ana_hbonds.csh script can also be run manually. For this simply copy the
ana_hbonds.csh and count_hbonds.awk scripts from the tool directory into
the analysis directory and type:
./ana_hbonds.csh hbonds.disp
Output files:
- nbcontacts.disp: this file contains a listing of all intermolecular hydrophobic contacts
over the ensemble of structures. It is automatically parsed by HADDOCK using the ana_hbonds.csh
script located in the tools directory. This scripts generate a listing (ana_nbconbtacts.lis)
of intermolecular hydrophobic contacts including the number of occurrences and the average C-C distance.
-
Note8: The ana_hbonds.csh script can also be run manually. For this simply copy the
ana_hbonds.csh and count_hbonds.awk scripts from the tool directory into
the analysis directory and type:
./ana_hbonds.csh nbcontacts.disp
Violations analysis
HADDOCK performs automatically a number of violations analysis, generating a listing of violations including the number of times a restraint is violated and the average distance and violation per restraint. This is done for distance restraints (all distances (distances + Hbonds), distances only, unambiguous distances only, ambiguous distances only, dihedral angle restraints). A number of .lis files are generated in the analysis directory:- ana_dihed_viol.lis: dihedral angles violations if a dihedral file has been input in the new.html
- ana_dist_viol.lis: all distance (including Hbonds) restraints violations
- ana_hbond_viol.lis: hydrogen bond restraints violations
- ana_noe_viol_all.lis: all distance restraints violations
- ana_noe_viol_unambig.lis: unambiguous distance restraint violations
- ana_noe_viol_ambig.lis: ambiguous distance restraints (this is the restraint type typically used for the ambiguous interaction restraints (AIRs).
Example:
Rexp= 2.000 Rave= 4.739 Viol= -2.739 #viol= 200 ( B 36 HIS N ...
Rexp= 2.000 Rave= 4.626 Viol= -2.626 #viol= 200 ( B 65 ASP N ...
Rexp= 2.000 Rave= 4.345 Viol= -2.345 #viol= 200 ( B 33 GLN N ...
Rexp= 2.000 Rave= 4.037 Viol= -2.037 #viol= 1 ( B 92 GLY N ...
Rexp= 2.000 Rave= 3.225 Viol= -1.225 #viol= 63 ( A 37 SER N ...
...
Rexp= 2.000 corresponds to the upper distance restraint (in Angstrom) defined in the AIR restraint file).
Rave= 4.739 corresponds to the average distance (in Angstrom) in the calculated structures.
Viol= -2.739 corresponds to the violation in Angstrom.
#viol= 200 corresponds to the number of structures in which the restraint is violated.
Manual analysis
An important part of the analysis, namely the analysis of the clusters, needs to be performed manually. A number of scripts are provided for this purpose in the runX/tools directory.To run it type:
$HADDOCKTOOLS/ana_structures.cshin the directory where file.list has been created (e.g. structures/it1 or structures/it1/water).
Ten files are created:
- structures_haddock-sorted.stat
- sorting based on haddock score
(as in file.list)
- structures_air-sorted.stat
- sorting based on distance restraint energy
- structures_airviol-sorted.stat
- sorting based on number of distance violations
- structures_bsa-sorted.stat
- sorting based on buried surface area
- structures_dH-sorted.stat
- sorting based on total energy difference calculated as total energy of the complex - Sum of total energies of the individual components
- structures_Edesolv-sorted.stat
- sorting based on desolvation energy calculated using the
empirical atomic solvation parameters from Fernandez-Recio et al. JMB 335:843 (2004)
- structures_ene-sorted.stat
- sorting based on total energy (only intermolecular components for vdw and elec energies)
- structures_nb-sorted.stat
- sorting based on non-bonded intermolecular energy
- structures_nbw-sorted.stat
- sorting based on weighted non-bonded intermolecular energy ( 1*vdw + 0.1*elec)
- structures_rmsd-sorted.stat
- sorting based on RMSD from best (lowest) HADDOCK score structure
#struc haddock-score RMSD-Emin Einter Enb Evdw+0.1Eelec Evdw Eelec Eair Ecdih Ecoup Esani Evean Edani #NOEviol #Dihedviol #Jviol #Saniviol #veanviol #Daniviol bsa dH Edesolv e2a-hpr_71w.pdb -164.13017 0.000 -629.446 -635.908 -107.853 -49.1804 -586.728 6.4629 0 0 0 0 0 0 0 0 0 0 0 1613.82 -8593.04 1.74954 e2a-hpr_171w.pdb -156.04058 0.748 -613.411 -624.683 -103.675 -45.7858 -578.897 11.2722 0 0 0 0 0 0 0 0 0 0 0 1663.99 -8501.99 4.3974 e2a-hpr_38w.pdb -150.756688 0.624 -574.337 -587.378 -97.1234 -42.6507 -544.727 13.0407 0 0 0 0 0 0 0 0 0 0 0 1688.07 -8600.72 -0.464658 ...
The first line of those files gives the description of the columns, e.g. the first column corresponds to the pdb file, the second column to the combined HADDOCK score, the third to the backbone RMSD from the lowest energy structure, the third column to the total intermolecular energy (sum of all energy terms), the fourth column to the intermolecular non-bonded energy (vdw+elec),...
You can generated a plot of the HADDOCK score as a function of the RMSD (using XMGR for example).
A simple script called make_ene-rmsd_graph.csh is provided in $HADDOCKTOOLS which allows you to generate an input file for Xmgr/XmGrace. Simply specify two columns to extract data from and a filename:
$HADDOCKTOOLS/make_ene-rmsd_graph.csh 3 2 structures_unsorted.statThis will generate a file called ene_rmsd.xmgr which you can display with xmgr or xmgrace:
xmgrace ene_rmsd.xmgr
cluster_struc is a simple C++ program provided in the tools directory that read the fileroot_rmsd.disp file containing the pairwise rmsd matrix and generates clusters. This program should have been compiled for your system during installation.
Two clustering algorithms are implemented:
- using an algorithm as described in Daura et al. Angew. Chem. Int. Ed. 38:236-240 (1999):
count number of neighbors using cut-off, take structure with largest number of neighbors with
all its neighbors as cluster and eliminate it from the pool of clusters. Repeat for remaining
structures in pool.
- full linkage: add a structure to a cluster when its distance to any element of the cluster is less than the cutoff.
The usage is:
cluster_struc [-f] fileroot_rmsd.disp cut-off min_cluster_size >cluster.outExample for its use:
cluster_struc e2a-hpr_rmsd.disp 7.5 4 >cluster.outwill create clusters using a 7.5 A RMSD cut-off requiring a minimum of four structures per cluster.
The output looks like:
Cluster 1 -> 8 1 2 3 5 6 7 9 10 11 12 13 14 15 ...
Cluster 2 -> 23 25 26 29 39 62 66 67 72 74 78 ...
Cluster 3 -> 153 4 32 43 96 131 147 158 163 ..
The numbers correspond to the structure number in the analysis file. For example
8 corresponds to structure number 8 in
analysis, i.e, the eigth structure in file.list in it1/water.
The first structure of each cluster above corresponds to the cluster center. The remaining structures are sorted according to
their index.
cluster_fcc.py is a python code provided in the tools directory that read the fileroot_fcc.disp file containing the pairwise fraction of common contact matrix and generates clusters. The clustering algorithm is described in Rodrigues et al. Proteins: Struc. Funct. & Bioinformatic, 80 1810-1817 (2012).
The usage is:
Usage: cluster_fcc.pyExample for its use:[options] Options: -h, --help show this help message and exit -o OUTPUT_HANDLE, --output=OUTPUT_HANDLE Output File [STDOUT] -c CLUS_SIZE, --cluster-size=CLUS_SIZE Minimum number of elements in a cluster [4]
python cluster_fcc.py e2a-hpr_fcc.disp 0.75 -c 4 >cluster.outwill create clusters using a 0.75 FCC cut-off requiring a minimum of four structures per cluster.
The output looks the same as for the RMSD-based clustering explained above
To run it, type with as argument the output file of the clustering, e.g.:
$HADDOCKTOOLS/ana_clusters.csh [-best #] analysis/cluster.out
The [-best #] is an optional (but recommended!) argument to generate additional files with cluster averages calculated only on the best # structures of a cluster. The best structures are selected based on the HADDOCK score defined in run.cns, i.e. the sorting found in file.list. This allows to remove the dependency of the cluster averages upon the size of the respective clusters. The following example will calculate cluster averages over the best 45 structures.
$HADDOCKTOOLS/ana_clusters.csh -best 4 analysis/cluster.out
The ana_clusters.csh script analyzes the clusters in a similar way as the ana_structures.csh script, but in addition generates average values over the structures belonging to one cluster. It creates a number of files for each cluster containing the cluster number clustX in the name:
- file.cns_clustX
- contains the name of all the pdb files that belong to the cluster X (CNS format)
- file.nam_clustX
- contains the name of all the pdb files that belong to the cluster X
- file.list_clustX
- contains the name of all the pdb files that belong to the cluster X (list format)
And in addition if the option -best Y is used:
- file.cns_clustX_bestY
- contains the name of the best Y pdb files that belong to the cluster X (CNS format)
- file.nam_clustX_bestY
- contains the name of the best Y pdb files that belong to the cluster X
- file.list_clustX_bestY
- contains the name of the best Y pdb files that belong to the cluster X (list format)
Note9: Those files can be used to repeat the HADDOCK analysis for a single cluster (see below).
- file.nam_clustX_bsa
- contains the buried surface area of each structure of cluster X
- file.nam_clustX_dH
- contains the total energy difference calculated as total energy of the complex - Sum
of total energies of the individual components
- file.nam_clustX_Edesol
- contains the desolvation energy calculated using the empirical atomic solvation parameters
from Fernandez-Recio et al. JMB 335:843 (2004)
- file.nam_clustX_ener
- contains all the energy terms (intermolecular, Van der Waals, electrostatic and AIR) for
each structures of cluster X
- file.nam_clustX_haddock-score
- contains the combined haddock score
- file.nam_clustX_rmsd
- contains the RMSD of each structure of cluster X from the best (lowest) HADDOCK score
structure of cluster X.
- file.nam_clustX_rmsd-Emin
- contains the RMSD of each structure of cluster X from the best (lowest) HADDOCK score
structure of all calculated structures
- file.nam_clustX_viol
- contains the number of AIR and dihedral violations per structure
Note10: The ordering of the structures in those files follows the HADDOCK score ranking.
- cluster_bsa.txt
- contains the average buried surface area of each cluster and the standard deviation
- cluster_dH.txt
- contains the average total energy difference calculated as total energy of the complex
- Sum of total energies of the individual components
- cluster_Edesolv.txt
- contains the average desolvation energy calculated using the empirical atomic solvation parameters
from Fernandez-Recio et al. JMB 335:843 (2004)
- cluster_ener.txt
- contains the average energy terms of each cluster and the standard deviations
- cluster_haddock.txt
- contains the average combined haddock score
- cluster_rmsd.txt
- contains the average RMSD and standard deviation from the best (lowest) HADDOCK score structure
of cluster of the structures belonging to that cluster
- cluster_rmsd-Emin.txt
- contains the average RMSD and standard deviation of the clusters from the
best (lowest) HADDOCK score structure of all calculated structures
- cluster_viol.txt
- contains the average AIR and dihedral violations for each cluster and the
standard deviations
- clusters_haddock-sorted.stat
- contains the various cluster averages sorted as a function of the
combined haddock score
- clusters.stat
- contains the various cluster averages sorted as a function of the cluster number
- clusters_air-sorted.stat
- contains the various cluster averages sorted accordingly to the AIR energy
- clusters_bsa-sorted.stat
- contains the various cluster averages accordingly to the buried surface area
- clusters_dani-sorted.stat
- contains the various cluster averages accordingly to the diffusion anisotropy restraint energy
- clusters_dH-sorted.stat
- contains the various cluster averages accordingly to the total energy difference calculated
as total energy of the complex - Sum of total energies of the individual components
- clusters_Edesolv-sorted.stat
- contains the various cluster averages accordingly to the desolvation energy calculated using
the empirical atomic solvation parameters from Fernandez-Recio et al. JMB 335:843 (2004)
- clusters_ene-sorted.stat
- contains the various cluster averages accordingly to the intermolecular energy (restraints+vdw+elec)
- clusters_nb-sorted.stat
- contains the various cluster averagesd accordingly to the intermolecular non-bonded energy (vdw+elec)
- clusters_nbw-sorted.stat
- contains the various cluster averages accordingly to the weighted intermolecular non-bonded energy (vdw+0.1*elec)
- clusters_sani-sorted.stat
- contains the various cluster averages accordingly to the RDC (direct, SANI) restraint energy
- clusters_vean-sorted.stat
- contains the various cluster averages accordingly to the RDC (intervector projection angles, VEAN)
restraint energy
You can plot the HADDOCK score of the clusters as a function of their RMSD from the lowest energy structure (using xmgr/xmgrace for example).
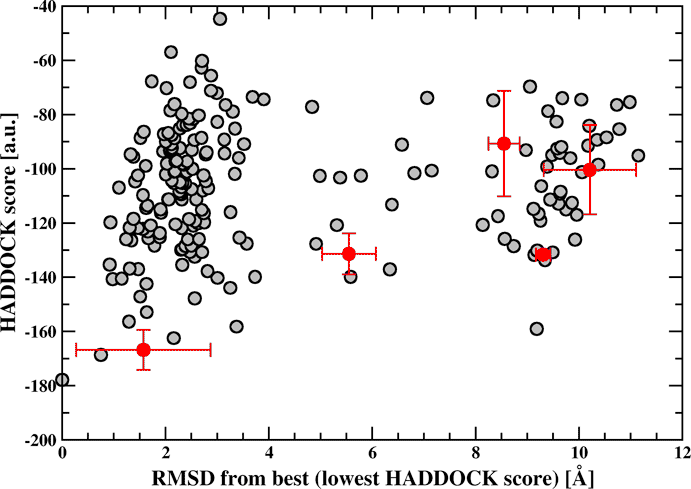
The gray circles correspond to the individual structures and the filled circles correspond to the cluster averages with the standard deviation indicated by bars.
The assumption is then that the best (lowest) HADDOCK score structures of the best (lowest) HADDOCK score cluster are the best solution generated by HADDOCK. It is then up to you to confirm that using any kind of information you can get such as for example:
- mutagenesis data
- conservation of given residues from multiple alignments
- ...
Rerunning the analysis for a given cluster
It is possible to rerun the HADDOCK analysis for a given cluster. For this, the file.cns, file.list and file.nam files should be renamed by adding for example a suffix _all. These three files contain the sorted list of all structures calculated. Similarly, the analysis directory should be renamed. Create then an empty analysis directory and cope the files containing the PDB file listings for a given cluster (these are created when performing the analysis of the clusters with ana_clusters.csh) to file.cns, file.list and file.nam, respectively.To simplify this entire procedure, we are providing a csh script named make_links.csh in the tools directory (defined by the environment variable $HADDOCKTOOLS). To make the links type:
$HADDOCKTOOLS/make_links.csh clust1
This will automatically move the original files (file.cns, file.list and file.nam) and rename
the analysis directory. A new analysis directory called analysis_clust1 will be created and
a link to it will be created as analysis. Similarly, links will be created for the three listing files:
file.cns -> file.cns_clust1 file.list -> file.list_clust1 file.nam -> file.nam_clust1To rerun the analysis go back to the runX directory and restart HADDOCK.
Warning: In case you wish to experiment with different clustering cut-offs restore first the original files containing the information for all calculated structures with the command:
$HADDOCKTOOLS/make_links.csh all