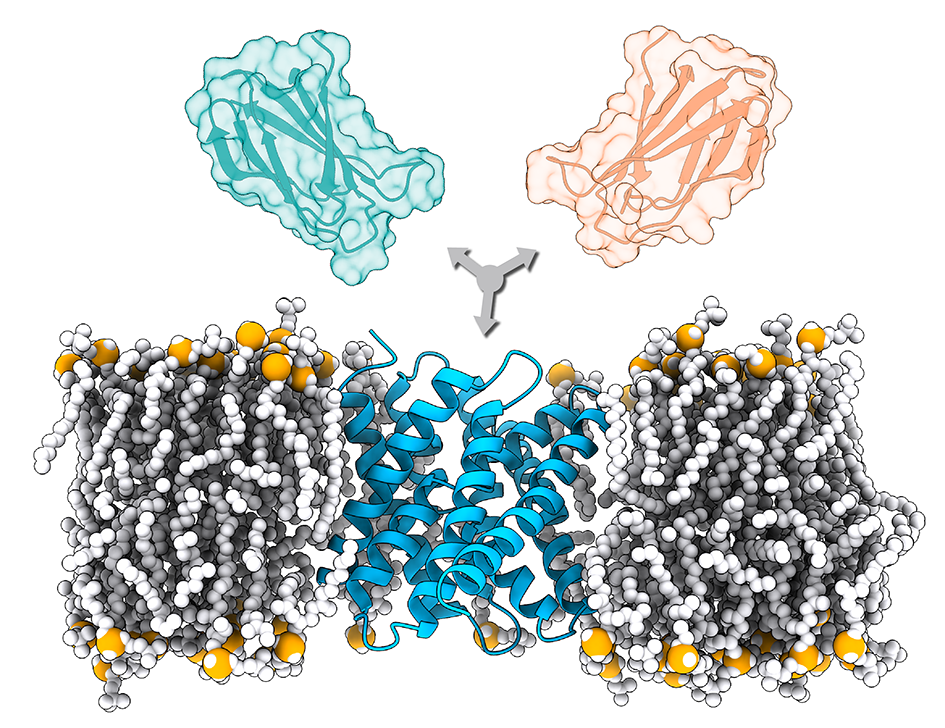
Membrane proteins are among the most challenging systems to study with experimental techniques in the structural biology field. The increasing number of deposited structures of membrane proteins has opened the route to modeling their complexes by methods such as computational docking. We have developed a new protocol for the modeling of membrane-associated protein assemblies, which successfully integrates a simulated membrane calculated by the MemProtMD database into a first docking step by LightDock with a flexible final refinement with HADDOCK. This protocol should shed light on the still dark fraction of the interactome consisting of membrane proteins and will be extremely useful for drug discovery, antibody design and personalized medicine.
Nature Comm. 11, 6210 (2020)
https://doi.org/10.1038/s41467-020-20076-5