

How to use information about interactions in HADDOCK?
Back to main best practice page

Best practice guide
As you probably saw in the previous step there are many ways how to obtain structures of molecules that you want to dock. The next step is to define the way you expect these molecules to interact. HADDOCK is an information-driven tool, which means that the more available information about binding you have, the more meaningful your results will be. Based on the available information we distinguish between following options:
- What information about binding is available?
- Getting restraints HADDOCK-ready
- Dos and Don’ts
- Complimentary software related to restraints for HADDOCK
What information about binding is available?
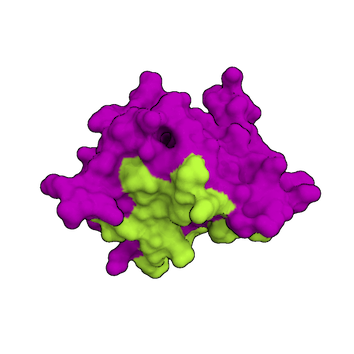
1.) Information about the interface is available
Unambiguous Interaction restraints
If your predictions are highly reliable and you wish to have all of them applied during docking, define them a unambiguous restraints. These can be for example template-derived pairwise distance restraints (tutorial), MS crosslink data (tutorial) or cryo-EM connectivity data (tutorial).
Ambiguous Interaction Restraints (AIRs)
Nevertheless, as in life, in science one also needs to be somewhat critical to the data one works with. If you are not 100% sure about the interaction information and want to be cautious while incorporating it into your docking, use ambiguous interaction restraints, unique for HADDOCK. Here, for each docking trial a fraction of these restraints will be randomly removed, which ensures a wider sampling satisfying always a different subset of predefined restraints. Thus, if some of the restraints are artificial, these can be filtered out if the complex satisfying them is unfavorable.
For AIRs, it is important to define the residues at the interface for each molecule based on experimental data that provides information on the interaction interface.
In the definition of those residues, one distinguishes between "active" and "passive" residues.
-
The "active" residues are of central importance for the interaction between the two molecules AND are solvent accessible. Either main chain or side chain relative accessibility should be typically > 40%, sometimes a lower cutoff might be used as well, for example the HADDOCK server uses by default 15%. Throughout the simulation, these active residues are restrained to be part of the interface, if possible, otherwise incurring in a scoring penalty.
-
The "passive" residues are all solvent accessible surface neighbors of active residues (<6.5Å). They contribute for the interaction, but are deemed of less importance. If such a residue does not belong in the interface there is no scoring penalty.
In general, an AIR is defined as an ambiguous intermolecular distance between any atom of an active residue of molecule A and any atom of both active and passive residues of molecule B (and inversely for molecule B).
Ambiguous distance restraints are described in the HADDOCK manual and more about parameters in the run.cns file is written here.
Using ambiguous restraints for docking is described in several tutorials: local installation tutorial, basic protein-protein tutorial, small molecule docking tutorial or antibody-antigen docking tutorial.
Other kinds of restraints
HADDOCK can utilize plenty of experimental information. Here we describe other types of restraints supported by HADDOCK:
2.) Information about the interface is not available
If there is no direct information about the interacting residues available, one can still browse through the available literature or employ bionformatic prediction to gain some information about the potential complex. HADDOCK offers a plethora of ways for these scenarios.
Information about the quaternary structure of proteins (symmetry)
Symmetry restraints
HADDOCK offers the possibility to define multiple symmetry relationships within or in between molecules. This is done by using symmetry distance restraints. By defining multiple pairs of distances between the CA atoms of two chains, various symmetries can be enforced. Symmetry restraints are described in the manual here.
Ab-initio multi-body docking with symmetry restraints is described this ab-initio tutorial.
Non-crystallographic symmetry restraints (NCS)
The NCS option imposes non-crystallographic symmetry restraints: It enforces that two molecules, a fraction thereof or even two sub-domains within the same molecule should be identical without defining any symmetry operation between them. Non-crystallographic symmetry restraints are described in the manual here.
Ab-initio multi-body docking with NCS restraints is described here.
Membrane Z-positioning restraints
These restraints do not deal with symmetry, but can be useful in guiding the docking of membrane proteins. This type of restraints is used to keep segments within or outside of a defined Z-coordinate range. They can be used for docking of membrane proteins but can be use generically as well.
They are described in the HADDOCK manual here.
Ab-initio docking
Random interaction restraints
HADDOCK offers to define random AIRs from solvent accessible residues (>20% relative accessibility) in case there is no experimental information. The sampling will be done from the defined semi-flexible segments. This can be useful for ab-initio docking to sample the entire protein surface. To ensure a thorough sampling of the surface, the number of structures generated at the rigid-body stage (it0) should be increased (e.g. 10000), depending on the extent of the surface to be sampled. These random restraints are described here.
Random interaction restraints are used in the binding site tutorial.
Surface contact restraints
Surface contact restraints can be useful in multi-body (N>2) docking to ensure that all molecules are in contact and thus promote compactness of the docking solutions. As for the random AIRs, surface contact restraints can be used in ab-initio docking; in such a case it is important to have enough sampling of the random starting orientations and this significantly increases the number of structures for rigid-body docking. They can be useful in combination with random interaction restraints definition (see above) or in refinement of molecular complexes. They are described in the manual here.
Center of mass restraints
Center of mass (COM) restraints are distance restraints that ensure close proximity of two molecules. Such restraints can be useful in multi-body (N>2) docking to ensure that all molecules are in contact and thus promote compactness of the docking solutions. Similarly to the contact surface restraints they can be useful in combination with random interaction restraints definition (see above) or in refinement of molecular complexes.
COM restraints are mentioned in multiple tutorials, for example: Refining the interface of the cryo-EM fitted models with HADDOCK, HADDOCK 2.4 CASP-CAPRI T70 ab-initio docking tutorial, Modelling a homo-oligomeric complex from MS cross-links.
Optimal settings for docking using bioinformatics predictions
When we are less certain about the of the interacting residues, it is better to enhance sampling by increasing the number of structures generated in each phase of docking.
| Parameter | run.cns name | default value | optimal value |
|---|---|---|---|
| Number of partitions for random exclusion | ncvpart |
2.0 | 1.1428 |
| Number of trials for rigid body minimisation | ntrials |
5 | 1 |
| Number of structures for rigid body docking (it0) | structures_0 |
1000 | 10000 |
| Number of structures for semi-flexible refinement (it1) | structures_1 |
200 | 400 |
| Number of structures for the final refinement (itw) | waterrefine |
200 | 400 |
| Number of structures to analyze | anastruc_1 |
200 | 400 |
IMPORTANT NOTE: The non-integer value of ncvpart can only be used in the web server. What the server is then doing is to pre-generate ambig.tbl_XXX files, where XXX indicated the model number. Those are then placed into the structures/it0 directory and noecv is set to false.
When such files are present, they will be read instead of the regular ambig.tbl. The automatic partioning into sets of restraints in CNS can only handle an integer number, meaning that the largest possible random removal is 50% (ncvpart=2). For more than 50% random removal custom ambig.tbl_XXX files must be generated prior to docking.
More about optimal settings for different docking scenarios can be found here.
Getting restraints HADDOCK-ready
In HADDOCK2.4 webserver active and passive residues can be entered manually or in a tbl file of ambiguous and unambiguous restraints.
Such restraints file can be generated using the Generate Restraints from haddock-restraints web service and can be further used when using HADDOCK locally, since it is already CNS-formatted.
Haddock tools a bunch of useful tool available on Github a bunch of useful tool available on github for use with local version of HADDOCK.
- contact-chain, contact-segID - programs to calculate all heavy atom interchain contacts within a given distance cutoff - useful to define active/passive residues based on a template structure
- passive_from_active.py, active-passive_to_ambig.py - these scripts will automatically calculate a list of surface residues from the PDB to filter out buried residues and create an ambiguous interaction restraints file based on the list of active and passive residues
- restrain_bodies.py, restrain_ligand.py - scripts that will keep multiple chains or ligands keep together during the flexible parts of docking
- validate_tbl.py - this script checks the correctness of your restraints (CNS format) for HADDOCK.
Use of the HADDOCK tools is also described in the local HADDOCK tutorial.
More information about distance restraints:
Dos and Don’ts
| Don't | Do instead |
|---|---|
| define the entire protein as active | define only key interacting residues as active, if they are not know, define the surface of one molecule as passive |
Complimentary software related to restraints for HADDOCK
In bonvinlab a number of complementary webservers have been developed to help users to reevaluate restraints.
CPORT
CPORT is an algorithm for the prediction of protein-protein interface residues. It combines six interface prediction methods into a consensus predictor.
Tutorials using CPORT:
DISVIS
DISVIS visualizes and quantifies the information content of distance restraints between macromolecular complexes.
Tutorial describing DisVis:
- DisVis tutorial
- HADDOCK2.4 tutorial for the use of MS crosslinks
- Integrative modelling of the RNA polymerase III apo complex
SPOTON
SPOTON determines Hot-Spot residues at protein-protein interfaces.
Any more questions about restraints for HADDOCK? Have a look at the HADDOCK bioexcel forum hosted by ![]() . There is a very high chance that your problem has already been addressed.
. There is a very high chance that your problem has already been addressed.