

How to prepare structures for HADDOCK?
Back to main best practice page

Best practice guide
First step in your docking protocol is to know which molecules you want to find a complex for. This might sound easy, however it can be quite tricky. This section explains where to find or model input structures, how to edit them and prepare them for HADDOCK:
Which structures are available?
Experimental structures
In the best case scenario there is an experimental structure available. All crystallographic, NMR or cryo-EM structures protein structures are deposited in protein data banks:
-
Worldwide Protein Data Bank wwPDB
-
Protein Data Bank in Europe PDBe
-
The Research Collaboratory for Structural Bioinformatics Protein Data Bank RCSB PDB
-
Protein Data Bank Japan PDBj
-
Biological Magnetic Resonance Data Bank BMRB
Sequence and homologous proteins
In case when there is no experimental structure available for molecules of proteins of interest, one can use proteins homologs as templates for protein modeling. There are multiple tools that help us to do so. Some online tools for homologue search are here:
Once one finds the protein homologues, some freely available software for homology model building are here:
- SWISS-MODEL
- this online tool can both look for homologous proteins and build a protein model
- MODELLER
- online version ModLoop for loop modeling
- local version for homology or comparative modeling of protein three-dimensional structures
Homology modeling using these tools is described in our tutorial here:
Modelling of peptides and mutations in proteins
- Point-mutations in HADDOCK are handled by changing the amino acid name and HADDOCK will fill the missing side chains atoms. This step is further described here and can be done using the pdb_mutate.py tool in haddock-tools.
Note that pdb_mutate.py will not create the new side-chain atoms (this is handled by HADDOCK). But if you prefer to have control of the side-chain conformation rather use tools like Pymol to introduce the mutation. This is even recommended in the case of a mutation to Histidine as the server can not automatically guess the protonation state if the side-chain is missing.
- Pymol is an almost irreplaceable tool in every-day life of a computational chemist. Pymol is often used in a number of HADDOCK tutorials for structure preparations as well as analysis of docking results.
- Pymol offers a lot of handy plugins which extend its usability, for example peptide-building ,some of them can be found here:
- Pymol offers an option to mutate residues and choose the side chain conformation manually.
- Modelling of peptides using Pymol modeling scripts is described here.
- Rosetta
- Rosetta, as well as plenty other online tools have now functionalities with which you can build peptides from their sequences.
- A list of modified amino acids supported by HADDOCK can be found here.
Modeling of small molecules
- OpenEye OMEGA
- OMEGA uses the SMILES strings as input to generate three-dimensional (3D) conformations of ligands. OMEGA was used by our group in previous rounds of the D3R challenge.
- license necessary
- RDKit
- open source chemoinformatics and machine learning software
-
to prepare topology and parameter files for the ligand in CNS format one can use:
-
the PRODGR server maintained by Daan van Aalten at Dundee University: https://prodrg2.dyndns.org
This server allows you to draw your molecule or paste coordinates and will return topologies and parameter files in various format, including CNS. You should turn on the electrostatic to obtain partial charges. -
the Automated Topology Builder (ATB) and Repository developed in the group of Prof. Alan Mark at the University of Queensland in Brisbane: https://compbio.biosci.uq.edu.au/atb
-
Preparation of small molecules for docking is further described in the frequently asked questions page.
Using Molecular Dynamics for generating multiple conformations
Proteins are not rock-solid and HADDOCK can handle flexibility of the interface to a certain extent. An elegant way how to account for larger conformational changes is ensemble docking of conformations generated by Molecular Dynamics (MD). There is a number of MD engines available for generating of conformations such as:
Examples of using MD for HADDOCK are shown here:
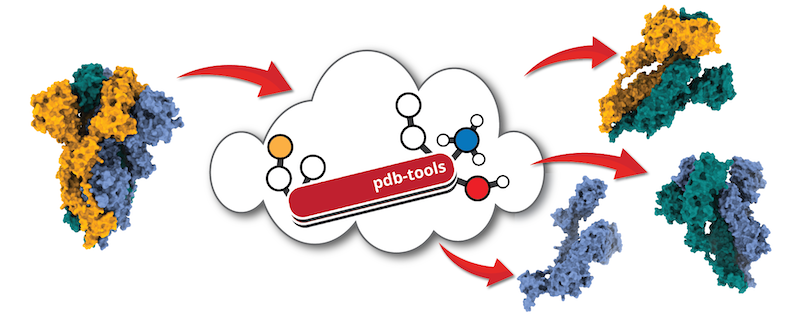
Editing pdb files
Upon acquiring the input structures provided you might want to modify in one way or the other. This might not be very straightforward since pdb files have to meet strict formatting requirements and are rather lengthy to edit manually. The HADDOCK group has therefore developed a pipeline called PDB-Tools where pdb files can be submitted and edited it as needed. PDB-tools are available here:
Tutorials:
Getting structures HADDOCK-ready
- Preparation of coarse-grained pdb files
- HADDOCK can now handle large complexes containing up to 20 chains. An elegant way how to increase the speed of these calculations is to use coarse graining with Martini.
-
Preparation of pdb files for the local version of HADDOCK2.4
-
Haddock tools are a bunch of useful tools available on Github that can be used to modify pdb or restraint files.
- A list of modified amino acids and another molecule types supported by HADDOCK can be found here.
Dos and Don’ts
| Don't | Do instead |
|---|---|
| submit a pdb file without checking it first | carefully inspect your pdb and remove any unwanted atoms (water molecules, ions, crystallization agents) |
| edit pdb files in Word, OpenOffice or LibreOffice editor | edit pdb files in an ASCII text editor |
| use residues with multiple occupancies (e.g. 124A, 124B) | use pdb_selaltloc.py to choose only one residue occupancy |
| use residues with overlapping numbering | use pdb_reres.py to renumber residues |
| use atoms with identical atom names | edit your molecule with an ASCII text editor to make all atom names unique or use pdb_uniqname.py from our PDB-tools |
| use a pdb file with incorrect formatting | pdb formatting is very strict, check your file with pdb_validate.py and reload and export the file in Pymol if necessary |
Once you have your structures HADDOCK-ready you can go to next step and define restraints.
Any more questions about pdb preparation for HADDOCK? Have a look at the HADDOCK bioexcel forum hosted by ![]() . There is a very high chance that your problem has already been addressed.
. There is a very high chance that your problem has already been addressed.